Глава 5. Гены и химики
За недолгие 15 лет генная инженерия превратилась в один из самых мощных инструментов познания живой материи. Получив возможность работать с индивидуальными генами, молекулярные биологи и генетики открыли совершенно новые их свойства. Именно благодаря генной инженерии обнаружено экзон-интронное строение генов эукариот, открыты многие регуляторные элементы, управляющие работой генов, выявлены мигрирующие гены. Возникла новая область молекулярной биологии - белковая инженерия: появилась возможность конструировать новые белки. Произошло это потому, что ученые могут иметь в своем распоряжении любой ген в индивидуальном состоянии и проводить изучение его структуры всеми доступными им методами. До недавнего времени специалисты работали лишь с механической смесью многих тысяч генов, к тому же разорванных случайным образом.
Как прочитать генетический текст. Первый вопрос, на который теперь можно получить ответ, - это первичная структура (или последовательность чередования нуклеотидных остатков) гена. Пятнадцать лет назад задача определения нуклеотидной последовательности сегмента ДНК длиной лишь в несколько десятков нуклеотидов (напомним, что средняя длина генов приближается к тысяче нуклеотидных остатков) не всегда была разрешимой. (Правда, с определением первичной структуры РНК дело обстояло лучше.)
Для РНК уже давно обнаружены ферменты - рибонуклеазы, которые расщепляли ее цепь специфически, т. е. после определенных остатков нуклеотидов (или, проще, по определенным буквам записанного в ней генетического текста). Так, рибонуклеаза TI, выделяемая из низшего гриба - аспергиллуса, разрезает любую РНК после остатков гуаниловой кислоты (т. е. после буквы G), а рибонуклеаза А из поджелудочной железы млекопитающих расщепляет РНК после пиримидиновых нуклеотидов (т. е. после букв U и С). Поэтому молекулу РНК можно постепенно расщеплять на все более и более мелкие перекрывающиеся фрагменты (вспомните частичное расщепление ДНК при построении их рестрикционных карт) до тех пор, пока не получались короткие олигонуклеотиды, доступные для химического анализа.
Первыми были исследованы транспортные РНК - самые короткие из известных тогда РНК (они состоят из 70-80 нуклеотидных остатков). В середине 60-х годов в лабораториях Холли (США) и А. А. Баева (СССР) их сначала научились получать в индивидуальном состоянии, а затем определили их нуклеотидную последовательность. Потребовалось еще 10 лет, чтобы определить нуклеотидную последовательность вирусной РНК длиной примерно в 3500 нуклеотидных остатков. Эта работа требовала огромного труда самых искуснейших экспериментаторов.
Для ДНК ферментов, подобных специфическим рибонуклеазам, известно не было. Но с открытием рестриктаз изучение нуклеотидной последовательности сдвинулось с места. Теперь появилась возможность некоторые из доступных индивидуальных ДНК (к ним относились ДНК мелких вирусов и бактериофагов) направленно разрезать на фрагменты, с этих фрагментов с помощью PHK-полимеразы получать комплементарные РНК-копии и наконец, определять нуклеотидную последовательность этих РНК.
Однако метод определения первичной структуры, как его часто называют секвенирования (от английского слова - последовательность) оставался прежним, очень медленным и сложным. Число же клонированных генов и их фрагментов росло. Требовались принципиально новые методы, которые бы в сотни раз ускорили анализ последовательности ДНК. И такие методы были разработаны.
Рассмотрим два из них, наиболее совершенных и распространенных. Первый называют методом Мак- сама - Гилберта, по имени его создателей известного американского ученого У. Гилберта и его сотрудника А. Максама. А. Максаму и У. Гилберту ничего не пришлось изобретать заново. Они "лишь" довели до совершенства идеи и разработки, по частям заложенные в работах многих предшественников: советских ученых С. К. Василенко, Е. Д. Свердлова, А. Д. Мирзабекова, бельгийца В. Фирса, англичанина Ф. Сэнгера и некоторых других. В чем суть этого метода?
Рестрикционный фрагмент двутяжевой ДНК длиной в несколько сот пар нуклеотидов метят радиоактивным фосфором по его 5′-концам. Делают это с помощью фермента полинуклеотид-киназы, который выделяют из клеток кишечной палочки, зараженной бактериофагом Т4. Этот фермент отщепляет фосфатную группу от молекулы АТФ и переносит ее на 5′-конец полинуклеотида. Затем двуспиральную ДНК денатурируют и отдельные цепи ДНК разделяют электрофорезом в геле. После разделения каждую из цепей отдельно вымывают из геля и в отдельности изучают.
Далее наступает самый важный момент анализа: надо расщепить ДНК избирательно, по каждому из четырех видов нуклеотидов, ее составляющих; причем сделать это следует так, чтобы в каждом случае каждая отдельная цепь ДНК расщепилась только один раз. На рисунке 32 показано, как эти требования выполняются на примере сравнительно короткой нуклеотидной последовательности ДНК. Заметим, что А. Максаму и Г. Гилберту не удалось найти условий избирательного расщепления ДНК по А и по Т. Но это не является препятствием, так как существуют условия, когда ДНК расщепляется только по G и только по С.
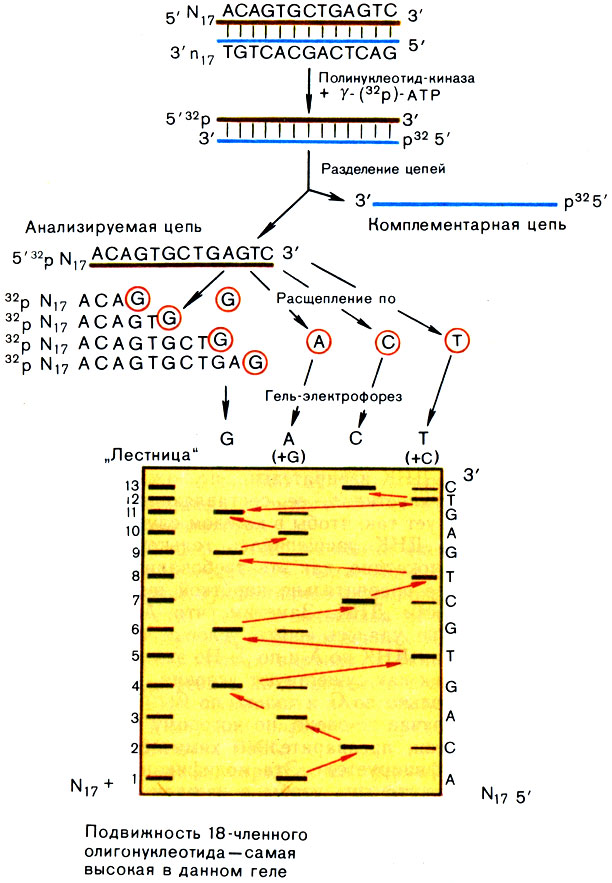
Рис. 32. Схема определения 13-членной нуклеотидной последовательности в 30-членном полинуклеотиде химическим методом Максама - Гилберта. Фрагменты короче 18 нуклеотидов 'ушли' за пределы геля
В каждом случае то звено, по которому проводится расщепление цепи, предварительно химически видоизменяется (модифицируется). Эта модификация как раз и должна проходить так, чтобы в каждом случае видоизменялась в среднем одна из букв на одну молекулу ДНК.
Из рисунка 32 видно, что препарат ДНК, разделенный на четыре порции, дает четыре набора фрагментов. Их фракционируют также электрофорезом в геле. На этот раз гель изготавливают из полиакриламида. Для этого в узкий промежуток между двумя стеклами заливают раствор акриламида вместе с некоторыми добавками. Полимеризуясь, акриламид образует длинные полимерные цепи, а добавки сшивают эти цепи в некоторых местах. В итоге получается полимерная сетка, которая набухает в воде и дает прозрачный бесцветный гель, напоминающий желе. Полимеризацию ведут в растворе электролита, содержащего денатурирующие агенты (обычно мочевину), которые не дают фрагментам ДНК слипаться во время электрофореза. Смесь фрагментов ДНК наносят на верхнюю часть геля (в специально изготовленные там лунки); гель соединяют с сосудами, где находятся электролит и электроды. Электрофорез проводят при высоком напряжении (1500-2000 В). Как и обычно, при электрофорезе в геле низкомолекулярные фрагменты движутся быстрее, чем более крупные. Электрофорез, который здесь применяется, настолько совершенен, что он позволяет разделить все фрагменты ДНК по их длине. Иными словами, отделить, скажем, фрагмент длиной в 91 нуклеотидный остаток от фрагментов длиной в 90 и 92 остатка. Это условие обязательное: без такой максимальной разрешающей способности определить последовательность ДНК нельзя. Кроме того, чем длиннее участок электрофореграммы, на котором в данном геле происходит разделение фрагментов с точностью до одного нуклеотида, тем длиннее сегмент ДНК, последовательность которого может быть определена в одном опыте. Многое, конечно, и здесь зависит от искусства экспериментатора, но определение последовательности участка ДНК длиной в 200-250 остатков в одном эксперименте никого сейчас не удивляет.
Остается добавить, что, как и обычно, зоны фрагментов ДНК выявляют на электрофореграмме с помощью авторадиографии (на гель в темноте накладывается рентгеновская фотопленка, которая засвечивается только в тех местах, где есть радиоактивная метка). На фотопленке видны лишь те фрагменты ДНК, которые помечены по 5′-концу, а расстояние, которое они прошли от верха геля, точно соответствует их длине. Теперь остается "прочитать" последовательность ДНК снизу вверх, каждый раз отыскивая ближайшую по горизонтам букву. Образец такой авторадиографии вы видите на рисунке.
Важно, что, просеквенировав вторую, комплементарную цепь фрагмента ДНК, исследователь имеет возможность проверить данные, полученные для первой цепи. Нужно лишь помнить, что G и Т в одной цепи соответствуют С и А в другой.
Второй метод секвенирования ДНК разработал замечательный английский ученый Ф. Сэнгер. На научном языке этот метод называют методом "с обрывом цепи" или "дидезокси"-методом. Прежде чем перейти к сути этого метода, скажем несколько слов о его создателе.
В 50-е годы Ф. Сэнгер прославился работами по определению полной аминокислотной последовательности белка-гормона инсулина. Это был первый случай, когда удалось расшифровать полную первичную структуру белка, причем всю методику исследований разработал сам Ф. Сэнгер. Пионерская работа Ф. Сэнгера, отмеченная Нобелевской премией по химии, породила целую серию расшифровок аминокислотных последовательностей белков.
В начале 1964 г. Ф. Сэнгер прочитал в Московском университете цикл лекций по структуре белков. Но свою последнюю лекцию он неожиданно для всех посвятил РНК, сообщив, что переключается на изучение нуклеиновых кислот. После этого он революционизировал методы секвенирования РНК, и первичная структура вирусной РНК, о которой мы упоминали, была определена его методом. Когда появилась возможность секвенировать большие ДНК, Ф. Сэнгер переключился на эту проблему. Он, в частности, нашел те условия электрофореза в полиакриламидном геле, которые позволяют разделять фрагменты ДНК, отличающиеся на один нуклеотидный остаток (их применили в своем методе А. Максам и У. Гилберт). Вклад Ф. Сэнгера в изучение структуры ДНК был так велик, что ему присудили вторую Нобелевскую премию по химии. До него только один исследователь - прославленная Мария Кюри была дважды удостоена этой высшей научной награды.
Многие детали методов Ф. Сэнгера и Максама - Гилберта сходны. Но есть и принципиальное отличие - секвенирование по Сэнгеру ведут с помощью ДНК-полимеразы, т. е. с помощью фермента, который не расщепляет, а синтезирует цепь ДНК. Этот принцип, несмотря на все последующие усовершенствования, до сих пор остался неизменным. В современном варианте, работая по методу Сэнгера, поступают следующим образом.
Сегмент ДНК, нуклеотидную последовательность которого собираются определить, встраивают в специальный вектор, изготовленный из ДНК бактериофага М13. Здесь следует вернуться к знакомой вам технике получения рекомбинантных ДНК.
Бактериофаг М13 необычный: он содержит одноцепочную кольцевую ДНК. Эта его особенность, как вы увидите дальше, и используется при секвенировании ДНК. Когда фаговая ДНК попадает в клетку кишечной палочки, она превращается в обычную двутяжевую кольцевую ДНК, которая реплицируется подобно другим фаговым или плазмидным ДНК. Значит, геном фага М13 можно иметь как в однотяжевом, так и в двутяжевом виде.
Из двутяжевой ДНК фага М13 сконструировали серию замечательных векторов. В ту ее область, которая не важна для репликации, встроили полилинкер с местами разрезания примерно десятком различных рестриктаз, а все остальные участки для тех же рестриктаз из ДНК убрали (таким образом, в полилинкере эти участки уникальны). Рядом с полилинкером встроили универсальную нуклеотидную последовательность из 17 нуклеотидных пар. В такой вектор после разрезания его одной из рестриктаз встраивают фрагмент ДНК, полученный посредством той же рестриктазы. Заметьте, что встроенный фрагмент оказался рядом с универсальной 17-членной последовательностью. Такой рекомбинантной ДНК заражают (трансфецируют) клетку. Рекомбинантная ДНК размножается в клетке и, одеваясь фаговыми белками, превращается в фаговые частицы. Однако в фаг попадает только одна из цепей, и, выделяя фаг, а затем фаговую ДНК, мы сразу получаем ее в однотяжевой форме.
Теперь можно непосредственно приступать к сек- венированию ДНК методом "обрыва цепи". Как мы говорили, в основе этого метода лежит ДНК-полиме- разная реакция. А для ДНК-полимеразы обязательно требуется затравка. Для этого и была вставлена рядом с секвенируемым фрагментом 17-членная последовательность. Она расположена справа от него, т. е. ближе к 3′-концу молекулы ДНК. Затравкой будет 17-членный олигонуклеотид, комплементарный этой последовательности.
Если олигонуклеотид-затравку связать с ДНК-матрицей, а затем добавить ДНК-полимеразу и дезоксинуклеозидтрифосфаты, то получим полную ДНК-копию нужного фрагмента. Заметьте, что в ДНК-полимеразной реакции каждое последующее звено цепи присоединяется к гидроксильной группе на ее 3′-конце.
Идея Ф. Сэнгера заключалась в том, что вместе с нормальными дезоксинуклеозидтрифосфатами (dNTP) в реакционную смесь вводили дидезоксинуклеозидтри- фосфат (ddNTP). Посмотрите внимательно на рисунок: остаток рибозы в ddNTR лишен гидроксилов как при 2′-, так и при 3′-атомах углерода. Если такой остаток попадет на 3′-конец растущей цепи, то и ДНК-по- лимераза не сможет присоединить к нему следующее звено, и в этой точке рост цепи остановится. Можно так подобрать соотношение dNTP и ddNTP, что остановка роста цепи будет происходить в среднем один раз на каждую растущую цепь. И тогда, проведя реакции полимеризации для каждого из четырех нуклеотидов, получим по сути дела такие же фрагменты, что и в случае метода Максама - Гилберта. Читая электрофореграмму (вернее, ее радиоавтограф), помните, что секвенируемый сегмент ДНК комплементарен синтезирующейся цепи (не забывайте переводить G в С, С в G и т. д., рис. 33).
Чтобы проиллюстрировать значение методов Максама - Гилберта и Сэнгера для современной молекулярной биологии и генетики, приведем такие цифры. В 1965 г. была известна первичная структура двух тРНК (общей длиной около 150 нуклеотидных остатков); к 1975 г., когда техника секвенирования РНК, казалось бы, достигла совершенства, удалось узнать первичную структуру более сотни различных РНК и фрагментов ДНК, общая длина которых составила около 10 000 нуклеотидных остатков; к 1986 г. была определена полная первичная структура многих вирусных геномов и плазмид, множества генов и регуляторных районов ДНК, суммарная длина которых превысила 6 млн. остатков. Сейчас известна полная первичная структура ДНК некоторых митохондрий и хлоропластов (эти клеточные структуры имеют собственную ДНК), причем в последнем случае длина молекул ДНК превышает 150 тыс. нуклеотидных пар.
Лучшее доказательство могущества современной генетической инженерии - это то, что в настоящее время идет серьезное обсуждение проекта секвенирования всего генома человека: ДНК общей длиной в 3×109 пар нуклеотидов! Конечно, технику секвенирования ДНК для решения такой грандиозной задачи (которую можно сравнить только с космическими проектами) потребуется усовершенствовать. Подсчитано, что самый опытный экспериментатор, занимаясь только секвенированием, может определить за год последовательность ДНК длиной в 100 ООО пар нуклеотидов. Это значит, что для реализации проекта секвенирования всего человеческого генома сейчас требуется 30 ООО лет. Однако ученые рассчитывают в ближайшие годы не менее чем в 100 раз увеличить скорость секвенирования ДНК. Уже появились первые модели приборов, которые могут определять последовательность ДНК автоматически.
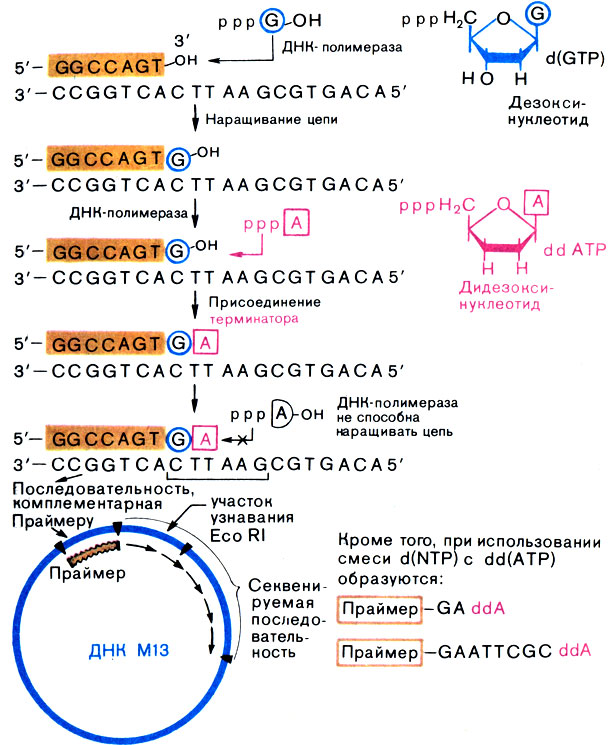
Рис. 33. Секвенирование ДНК терминаторным методом Сэнгера
Огромная информация, получаемая генными инженерами о нуклеотидной последовательности ДНК и РНК, может храниться только в памяти мощных ЭВМ. И только на ЭВМ эту информацию можно обработать. Компьютер может найти в гене промоторный участок, выявить ту его часть, в которой закодирован белок, пользуясь генетическим кодом, написать аминокислотную последовательность белка. Вот почему определение структуры генов привело одновременно к расшифровке первичной структуры множества белков, в том числе и тех, которые содержатся в клетке в ничтожных количествах и прямо изучены быть не могут. С помощью ЭВМ можно также сравнивать структуру разных генов, определять степень их схожести и на этой основе решать вопросы эволюции биологических видов. Одним словом, компьютер для современного генетика становится таким же необходимым прибором, как и аппарат для электрофореза.
Белковая инженерия. Техника рекомбинантных ДНК и расшифровка нуклеотидных последовательностей генов позволили начать работу по конструированию новых белков, которых нет в природе. Такое конструирование преследует минимум две цели. Во-первых, оно позволяет понять, как работают те или иные белки. Во-вторых, с его помощью можно получать более совершенные белки (более активные, более стабильные и т. д.).
С одним примером получения нового белка - химеры фермента β-галактозидазы и гормона соматостатина - мы уже встречались. Химеры, как вы помните, получают, сшивая гены разных белков друг с другом. Можно представить трудность создания белка со многими взаимодополняющими активностями, что-то вроде молекулярного комбайна.
Здесь, однако, сами сшиваемые гены остаются без изменений. Тем не менее мы располагаем теперь способами изменять любой нуклеотидный остаток в гене (т. е. проводить его направленный мутагенез). Такие замены в кодирующих участках генов, естественно, приводят к заменам одних аминокислот в белке на другие. Если такие замены делать по заранее разработанному плану, то в итоге можно получать белки с совершенно новыми свойствами.
Чтобы осуществить такие направленные замены, ген белка помещают в вектор, изготовленный из ДНК фага М13, точно так же, как и перед секвенированием его по методу Сэнгера. Затем к выбранному участку гена (в котором закодирован интересующий нас участок белка) синтезируют олигонуклеотид длиной в 18-20 остатков (олигонуклеотид такой длины прочно свяжется потом с геном). Этот олигонуклеотид комплементарен гену по всей длине, кроме той буквы, которую можно заменить. На рисунке он специально обозначен звездочкой. Этот олигонуклеотид используют как затравку для ДНК-полимеразы. При этом ДНК-матрицу с видоизменяемым геном используют в однотяжевой форме. ДНК-полимераза синтезирует на матрице новую цепь. В итоге получается двуспиральная ДНК с одним дефектом - после репликации получается ДНК как с исходным геном, так и с мутированным геном. Эти последние ДНК "вылавливают" из смеси, пользуясь тем же олигонуклеотидом как зондом (ход клонирования генов с помощью зондов мы подробно описали в предыдущей главе). Этот зонд будет лучше связываться с мутированным геном, чем с исходным, и можно найти такие условия, когда он будет вылавливать только рекомбинантные ДНК с мутированным геном. В результате экспрессии мутированного гена получают видоизмененный белок с запланированными заменами аминокислот (рис. 34).
Весь этот подход назвали олигонуклеотид-направляемый мутагенез генов. Создали его совсем недавно, но уже есть первые очень обнадеживающие результаты. Так, удалось сконструировать и получить фермент аминоацил-тРНК-синтетазу (тот самый, который специфически связывает аминокислоту с тРНК; см. главу 2), более активный, чем клеточный фермент. Удалось по заранее разработанному плану превратить белок-репрессор, регулирующий активность одного гена, в регулятор экспрессии другого гена. Все это только первые успехи. А возможности конструирования белковой инженерии неограничены (отметьте, белковая инженерия здесь продолжение генной). Вы можете спросить: зачем это надо? Неужели в огромном запасе генов биосферы не найдется нужного гена, чтобы создавать искусственный? Оказывается, возможности биологической эволюции ограничены. Каждая мутация происходит с довольно низкой вероятностью (в среднем 10-5). Допустим, чтобы изменить белок в нужную сторону, в гене должны произойти одновременно три мутации, каждая из которых организму невыгодна (ухудшает белок), а все вместе дают желаемый эффект. Идти путем накопления мутаций нельзя: гены с одной или двумя заменами дают неэффективный белок, который может привести к смерти организма. А вероятность наступления сразу трех равна 10-5 ⋅ 10-5 ⋅ 10-5= 10-15. Такие ничтожные вероятности в природе практически не реализуются, времени существования Земли на них просто не хватит. А искусственный, управляемый человеком мутагенез обходит эту трудность.
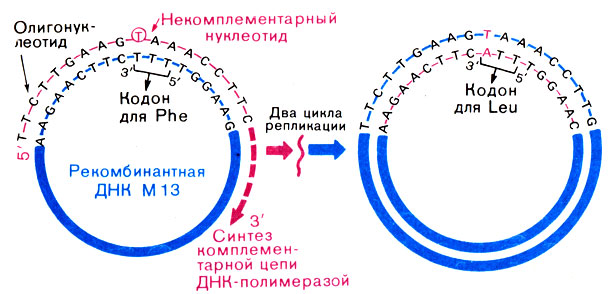
Рис. 34. Схема олигонуклеотид-направляемого мутагенеза гена, встроенного в однотяжевую ДНК фага М13
Затруднение в другом. Как правило, мы не знаем, как изменятся свойства белка, если в нем изменить один аминокислотный остаток на другой. Можно предсказать, например, что замена в гемоглобине в определенном положении глутаминовой кислоты на валин будет ухудшать растворимость белка, и мутантный гемоглобин, легко выпадающий в осадок, будет работать хуже. А то, что такой гемоглобин (серповидноклеточный гемоглобин S) окажется ядовитым для малярийного плазмодия и что человек-мутант по этому гену не будет болеть малярией, ни один химик-белковик предсказать не сможет. Или лучше сказать, пока не сможет.
Химический синтез генов. Для решения многих задач генной и белковой инженерии требуются синтетические олигонуклеотиды, будь то полилинкеры для сшивания генов, зонды для "вылавливания" генов при их клонировании или затравки с дефектами при направленном мутагенезе. Здесь мы хотели бы рассказать о том, как проводится синтез таких олигонуклеотидов, что может сделать сейчас химик-синтетик в области создания генетического материала.
С точки зрения органической химии не только олигонуклеотид, но и мононуклеотид - сложные соединения. В него входят компоненты, относящиеся к различным классам органических (гетероцикл, остаток сахара) и неорганических (остаток фосфорной кислоты) соединений. Нуклеотиды содержат множество различных по свойствам функциональных групп. Именно эти группы, столь необходимые для создания правильной макромолекулярной структуры нуклеиновых кислот, обеспечивающие комплементарность полинуклеотидных цепей, представляют наибольшую трудность при химическом синтезе олигонуклеотидов.
Задача такого синтеза, на первый взгляд, выглядит просто: нужно шаг за шагом (в том порядке, который задан нуклеотидной последовательностью олигонуклеотида) связывать фосфатную группу одного нуклеотида с гидроксильной группой другого так, чтобы каждый раз образовывалась 3′-5′-фосфодиэфирная связь. Сам по себе остаток фосфорной кислоты с гидроксилом сахара реагировать не будет; как сказали бы химики, он недостаточно реакционноспособен, его нужно дополнительно активировать. Это значит, что к фосфорной группе нужно направленно (не затронув другие функциональные группы нуклеотида) присоединить активирующую группировку. Далее реакцию активированного фосфата нужно прицельно провести с "правильной" ОН-группой сахара. Но по свойствам эта группа так похожа на функциональные группировки в других частях мононуклеотида, что единственный выход - как-то выключить эти группы из "игры". Приходится их защищать, т. е. навешивать на них защитные группировки, делая недоступными для реакций с активированной фосфатной группой.
Сильно упрощенная схема такого синтеза приведена на рисунке. Понятно, как много операций должен совершить химик-органик, чтобы получить олигонуклеотид всего из двух звеньев. Причем эти операции должны монотонно повторяться для присоединения каждого нового звена. К тому же в конце синтеза олигонуклеотид нужно освободить от всех защитных группировок. Возникла мысль: нельзя ли все эти операции автоматизировать?
Упорная многолетняя работа завершилась успехом. Автоматические синтезаторы олигонуклеотидов появились во многих лабораториях. Прибор, созданный в нашей стране в год сорокалетия Победы над фашизмом, получил название "Виктория-4". В этом названии звучат отзвуки победы, одержанной учеными над сложной научно-технической проблемой, а также имя одного из пионеров автоматического синтеза олигонуклеотидов В. К. Потапова. В. К. Потапов и З. А. Шабарова одними из первых сумели разработать так называемый твердофазный синтез олигонуклеотидов. Он и положен в основу работы автоматического синтезатора.
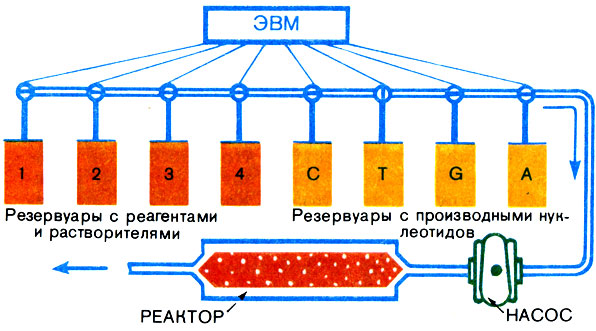
Рис. 35. Схема автоматического синтезатора олигонуклеотидов
Главный элемент автоматического синтезатора олигонуклеотидов - небольшой реактор - трубочка, заполненная полимером, приготовленным в виде мелких шариков (зерен). С зернами полимера химически сшит первый нуклеотид, фосфатная группа которого активирована. Таким образом, этот нуклеотид готов присоединить к себе следующий. Работой автомата управляет небольшая ЭВМ (рис. 35). По ее сигналу в реактор вводят раствор второго нуклеотида со всеми необходимыми реагентами. В память ЭВМ заложена нуклеотидная последовательность синтезируемого олигонуклеотида. Поэтому она "знает", какой из четырех нуклеотидов отправить в реактор. Если, например, эта последовательность АСТТСС, то к полимерному носителю с пришитым к нему А первым будет направлен С. Произойдет образование первого динуклеотида (АС). Далее по сигналу ЭВМ все ненужные реагенты отмываются, а фосфорная группа приводится в боевое состояние - активируется. В реактор вводится Т, и все начинается сначала.
Так шаг за шагом автомат синтезирует весь олигонуклеотид. Когда синтез закончен (в нашем случае после присоединения С), ЭВМ дает сигнал снять все защитные группировки (для этого в реактор подаются необходимые реагенты), а затем и снять сам олигонуклеотид с полимерного носителя (рис. 36). После дополнительной очистки олигонуклеотид готов к употреблению.
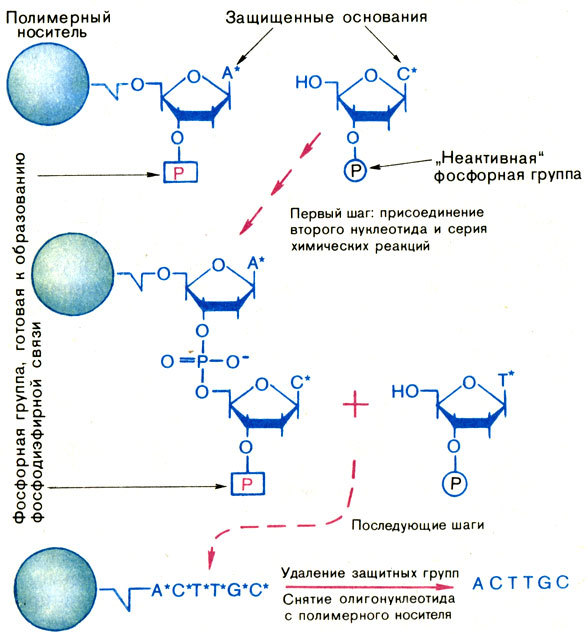
Рис. 36. Схема твердофазного синтеза олигонуклеотидов
На автоматическом синтезаторе олигодезоксирибонуклеотид длиной в 15-20 звеньев можно получить за несколько часов, а еще не так давно химик-синтетик тратил на это годы работы. Как вы уже знаете, олигонуклеотиды такой длины дают стабильные комплексы с комплементарными им участками ДНК и РНК; поэтому обычно их размеры устраивают генных инженеров. А если нужно синтезировать большой фрагмент гена или даже целый ген? И такие задачи сейчас выполнимы.
Первый синтетический ген получил знаменитый индийский ученый Г. Корана, работающий в США. Г. Корана - один из создателей современной химии нуклеиновых кислот, автор многих способов синтеза олигонуклеотидов. Ген, синтезированный Г. Кораной и его сотрудниками, по нынешним меркам невелик - около 150 нуклеотидных пар. Это ген одной из тРНК. Таким образом, он должен содержать в себе всю последовательность предшественника этой тРНК (но, конечно, в виде дезоксирибонуклеотида), к которой спереди присоединен промотор, а сзади - терминатор, т. е. сигналы начала и окончания транскрипции.
Эта работа была выполнена в конце 60 - начале 70-х годов, когда только зарождались современные способы химического синтеза генов. Автоматические синтезаторы олигонуклеотидов еще не существовали; поэтому первый ген мог быть синтезирован только усилиями многих ученых: в те годы лаборатория Г. Кораны была настоящим международным центром синтетической химии нуклеиновых кислот (работали в нем и советские ученые).
Одним из главных достижений Г. Кораны было создание блочного способа синтеза длинных полинуклеотидных последовательностей. Суть его показана на рисунке 37. Мы специально привели уже знакомый вам ген гормона соматостатина. Как видите, здесь опять используется принцип комплементарности нуклеиновых оснований. Каждый блок синтезируется так, чтобы его левая половина была комплементарна одному олигонуклеотиду, а правая - другому. После сборки всего гена из блоков их сшивают ДНК-лигазой. На концах синтетического гена обычно оставляют однотяжевые последовательности для встраивания его в вектор, разрезанный рестриктазами.
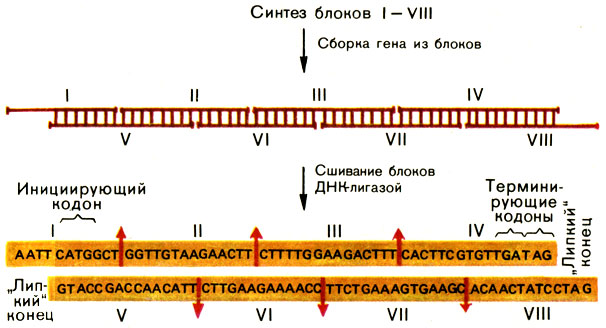
Рис. 37. Сборка синтетического гена гормона соматостатина
Блочный метод в сочетании с современными способами синтеза олигонуклеотидов позволил получить многие гены, в том числе такие большие белки, как интерфероны. Теперь химик-органик может создать наследственное вещество, что является замечательным доказательством власти современной науки над геном!
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'