9.1. Применения генетики человека
9.1.1. Генетическое консультирование
([71; 90; 101; 129; 136; 149; 205; 2258; 2293; 2323а; 2351])
Генетическое консультирование стало важной областью прикладной генетики человека: все больше людей обращается за советом к генетикам, и врачи все чаще посылают своих пациентов на консультацию по поводу диагноза наследственной болезни, её характера и риска рецидива. Поскольку средства массовой информации и медицинская литература распространяют все больше новых сведений по генетике, общественный и медицинский интерес к наследственным болезням постоянно нарастает. Что собой представляет генетическое консультирование? К генетическому консультированию относят следующие виды деятельности: а) установление диагноза; б) оценка риска повторных случаев; в) сообщение пациенту и его семье о вероятности повторных случаев; г) предоставление информации и благожелательное консультирование по многим вопросам, возникающим в связи с заболеванием, включая медицинский, экономический, психологический и социальный аспекты; д) предоставление информации, на основании которой консультируемому предстоит решить вопрос о целесообразности родов (включая результаты пренатальной диагностики).
Круг вопросов, подлежащих выяснению при генетическом консультировании, охватывает широкую область. Хотя в случае некоторых заболеваний экспертиза может иметь особенности, как правило, она стандартна. Оказывается, что только 30-50% обращающихся за консультацией семей имеют нарушения классического типа (моногенные болезни или хромосомные аберрации). Гораздо чаще консультации касаются различных врожденных пороков, умственной отсталости, задержанного развития, дизморфий, низкорослости и других отклонений, которые могут и не иметь генетической этиологии.
Генетическое консультирование обычно проводится врачами-специалистами. Многие врачи во всем мире специализируются теперь по медицинской генетике. В Соединенных Штатах Америки возникла новая профессия - "ассистент-генетик". Как правило, это женщины, которые обучались по специализированной двухгодичной университетской программе. Они работают вместе с врачами в медико-генетических клиниках и проводят большую часть до- и после-клинических посещений для сбора информации, консультирования и последующего наблюдения. Их участие в деятельности генетических служб приносит большую пользу.
Генетическое консультирование не преследует "евгенических" целей, оно имеет строго медицинскую направленность. Большинство исследователей считает, что нельзя давать супружеским парам советы относительно рождения детей, основанные на евгенических соображениях, даже в том случае, если результатом принятого ими решения может стать увеличение генетического бремени популяции. У пар, обращающихся за советом, поддерживают решимость сделать выбор самостоятельно; их решение никак не должно зависеть от возможных вредных последствий для генофонда популяции. Такой порядок твердо определяет место генетического консультирования в медицинской практике, для которой центром внимания, объектом советов и лечения являются отдельный человек и его семья, а не популяция в целом. К счастью, большинство пар выбирает тот путь, который соответствует благоприятному воздействию на генофонд популяции (т. е. предпочитает отказаться от деторождения в случае высокого риска).
Диагноз. Важно установить точный диагноз наследственного заболевания, используя все средства современной медицины. Точной постановке диагноза придается особое значение потому, что сходные фенотипы могут характеризоваться разными типами наследования или даже не быть врожденными. В случаях когда окончательный диагноз не вполне ясен, важную роль может сыграть семейный анамнез, поскольку четкий тип наследования, характерный, например, для аутосомно-доминантных признаков, может обеспечить основу для консультирования. При установлении точного диагноза часто оказываются полезными медицинские данные, зарегистрированные ранее. Поскольку многие наследственные болезни связаны с характерными чертами лица, может оказаться полезным знакомство с фотографиями членов семьи. Для диагностики сложных врожденных пороков (см. разд. 2.2.2) часто бывает необходимо исследование хромосом. Из-за того, что многие наследственные дефекты встречаются редко, даже специалистам в данной области медицины иногда трудно поставить точный диагноз. Диагностика и лечение генетических и связанных с ними заболеваний стали теперь новой отраслью - клинической генетикой. Одинаково хорошо знать все наследственные заболевания в каждом разделе медицины очень трудно, поэтому специалисты должны знакомиться с современными монографиями и руководствами [13; 85; 133; 187; 231], помогающими поставить соответствующий диагноз. В этом плане полезен "Каталог менделевских признаков человека", составленный Мак-Кьюсиком [133], однако этот материал необходимо дополнить обзорами, посвященными заболеваниям, наследующимся не по менделевским законам. Для клинической генетики особенно важна хорошая библиотека и умение работать с текущей литературой. Из-за быстрого роста знаний изучение журнальных статей, а не учебников и монографий для клинической генетики много важнее, чем для других областей медицины.
Родители, у которых дети рождаются мертвыми или умирают в младенческом возрасте, часто обращаются за генетической консультацией по поводу риска повторных случаев. О специфической патологии обычно в случае мертворождений мало что известно, так как патологоанатомическое или другие диагностические исследования часто не проводятся. Рекомендуется во всех случаях мертворождений или ранних неонатальных смертей проводить как минимум аутопсию, фотографирование, рентгенографию и бактериальный посев для установления диагноза, поскольку он необходим для генетического консультирования [71]. Если макроскопическая аутопсия не выявляет отклонений от нормы, патогистологическое или хромосомное исследование вряд ли даст сведения, имеющие диагностическое значение. Однако такие исследования оказываются необходимыми, если на аутопсии обнаруживаются множественные аномалии.
Даже опытные специалисты часто не могут поставить окончательный диагноз. Причина этого - необыкновенная сложность развития и его возможные нарушения за счет известных или по большей части неизвестных генетических, эпигенетических и средовых факторов. Диагностика моногенных заболеваний сопряжена с меньшей неопределенностью, чем различные врожденные пороки. Однако и в этой области, если судить по расширению каталога Мак-Кьюсика за последние годы (от 866 специфицированных локусов в 1971 г. до 1826 в 1985 г.),* сведения накапливаются очень быстро.
* (И до 4000 в 1988 г. - Прим. ред.)
Чтобы не отставать от бурного роста знаний, несколько исследовательских групп ввели информацию о клинических данных при генетических заболеваниях и врожденных пороках в компьютеры и разработали программы, позволяющие поставить диагноз с использованием этой информации. Хотя такой подход и оказался полезным в некоторых случаях, не все клинические генетики убеждены в преимуществах компьютерной диагностики. Ведь многие клинические признаки почти неуловимы и с трудом описываются словами. Однако разработки в этой области продолжаются, и со временем компьютерные программы, вероятно, будут пригодны для клинической работы.
Риск повторных случаев. Риск для заболеваний, наследующихся по Менделю, четко определяется и зависит от специфического типа наследования (табл. 9.1). Фактический риск для пациента при аутосомно-доминантном наследовании, например, зависит от степени пенетрантности и экспрессивности и от времени проявления многих заболеваний. Однако пациенты больше интересуются фактическим риском болезни с клиническими симптомами, чем формально-генетическим риском как таковым. Для заболеваний со сниженной пенетрантностью фактический риск будет меньше, чем формальный риск наследственной передачи. Например, при аутосомно-доминантном заболевании с 70%-ной пенетрантностью риск для потомства составит 35% (0,5 × 0,7 = 0,35), а не 50%. Для заболеваний с поздним проявлением риск уменьшается в случае, если человек здоровым переживает тот возраст, в котором должны обнаруживаться первые признаки болезни. Исчерпывающие данные относительно начала заболевания существуют, например, для хореи Гентингтона (14310), и их можно использовать для более точной оценки специфического риска [878] (см. рис. 3.4 и 9.1, приложение 8). По мере расшифровки генетической карты человека все большее число генетических дефектов можно будет диагностировать путем исследования расщепления тесно сцепленных генов-маркеров. Изменения ДНК, в особенности некодирующей ее части, происходят, по-видимому, по всему геному, и мутантная ДНК почти всегда обнаруживается с помощью генно-специфических зондов (разд. 2.3). Применение этого метода требует знаний о молекулярной структуре мутантного гена, вызывающего заболевание (табл. 9.2, А). Было обнаружено также сцепление локусов, определяющих болезни, с "анонимными" ДНК-маркерами, что может оказаться полезным для клиники (табл. 9.2,5). Попытка установить сцепление "анонимного" ДНК-маркера с геном болезни не требует знания сущности мутации, вызывающей наследственное заболевание. Однако если местоположение локуса болезни на хромосоме не определено и невозможно выявить ДНК-маркер этого участка, то шанс обнаружить такое сцепление невелик. Однако по мере все большего насыщения генной карты человека различными ДНК-маркерами, этот подход будет приносить все больше успехов.

Таблица 9.1. Повторный риск для родственников пробандов в случае редких менделирующих заболеваний
1 (Риск для детей пробандов с распространенными аутосомно-рецессивными заболеваниями зависит от частоты гена (самый высокий риск (4%), характерен для серповидноклеточной анемии (0,08 × 0,5)).)
2 (Риск незначительный, если заболевание вызвано новой мутацией. Недавно проведенное исследование указывает на то, что доля новых мутаций может быть намного меньше, чем ожидаемые 33% при сцепленных с Х-хромосомой заболеваниях с летальным исходом (за исключением миопатии Дюшенна) [2396]. (Оценки риска для сцепленных с Х-хромосомой рецессивных заболеваний используются, только если болезнь семейная, и не относятся к тем случаям, когда статус носительства у матери обусловлен новой мутацией.))
![Рис. 9.1. Кумулятивные кривые возраста начала (______) и риска проявления (----) заболевания для еще не заболевших сына или дочери пробанда с болезнью Гентингтона [2308]](pic/000078.jpg)
Рис. 9.1. Кумулятивные кривые возраста начала (______) и риска проявления (----) заболевания для еще не заболевших сына или дочери пробанда с болезнью Гентингтона [2308]
Таблица 9.2. Диагностика (включая пренатальную) генетических заболеваний с помощью тесно сцепленных ДНК- (и других) маркеровА. Определение с помощью ген-специфичных зондов* Серповидноклеточная анемия (АР) α- и β-талассемия (АР) Гемофилия А (сХР) Гемофилия В (сХР) Синдром Леша-Найхана (сХР) Семейная гиперхолестеринемия (АД) Фенилкетонурия (АР) Недостаточность α-антитрипсина (АР) Недостаточность антитромбина III (АР) Недостаточность гормона роста (АР) Б. Определение с помощью "анонимных" ДНК- маркеров, сцепленных с геном заболевания** Хорея Гентингтона (АД) Миопатия Дюшенна и Беккера (сХР) Миотоническая дистрофия (АД) Гемофилия А (сХР) Умственная отсталость, сцепленная с Х-хромосомой (сХ) Ретиносшизис (сХР) Поликистоз почек (АД)
АД - аутосомно-доминантный; АР - аутосомно-рецессивный; сХР - сцепленный с Х-хромосомой, рецессивный; сХ-сцепленный с Х-хромосомой.
* (Кроссоверы крайне маловероятны, т. к. маркеры ДНК, обнаруживаемые специфичными для гена зондами, тесно сцеплены и чрезвычайно близко расположены к участку мутации внутри или в окрестностях мутантного гена.)
** (Рекомбинация между маркерами ДНК и геном заболевания зависит от расстояния между ДНК-маркером и локусом болезни (см. разд. 3.4).)
Применение для диагностики метода сцепленных маркеров вместе с ДНК-зондами, специфичными для гена или хромосомы, предполагает, что мутантная ДНК присутствует в исследуемой семье и что число членов семьи, доступных для анализа, достаточно для того, чтобы четко установить, в каком положении (цис- или транс-) находится локус болезни по отношению к гену-маркеру и нормальному аллелю. Здесь может встретиться ряд трудностей. Например, было показано, что структура семей с болезнью Гентингтона [2307] в большинстве случаев не позволяет поставить преклинический диагноз на основе метода сцепленных маркеров, поскольку подходящих носителей мутации либо уже нет в живых, либо они еще слишком молоды для проявления заболевания, и поэтому их невозможно использовать для определения взаимного расположения гена-маркера и гена болезни.
Метод сцепления полезен только в тех случаях, когда диагноз невозможно поставить с помощью традиционных методов или когда они не дают определенного диагноза. Так, этот метод неоценим для пренатальной диагностики, для преклинического диагноза заболеваний с поздним началом или для определения носителей сцепленных с Х-хромосомой болезней и (в меньшей степени) носителей аутосомно-рецессивных патологий. Напротив, анализ ДНК-маркеров не используется при постнатальной диагностике таких заболеваний, как серповидноклеточная анемия, гемофилия или фенилкетонурия, при которых можно сделать соответствующий биохимический анализ крови на наличие генных продуктов и не нужно проводить семейного исследования. Изредка, в тех случаях, когда известна природа мутации, можно поставить диагноз с помощью прямого анализа ДНК. Вследствие генетической гетерогенности, т. е. из-за того, что одно и то же заболевание могут вызывать разные мутации, этот подход ограничен и не может применяться, пока исследуемая мутация точно не определена. Мы уже знаем, что со многими мутациями связаны, например, гемофилия А и семейная гиперхолестеринемия [685], но не серповидноклеточная анемия или недостаточность а-антитрипсина [43].
Генетическое консультирование при мультифакториальных заболеваниях (МФЗ), таких, как врожденные пороки развития, широко распространенные заболевания среднего возраста и эндогенные психозы, остается недостаточно точным (по сравнению с менделирующими заболеваниями), так как число генов и их относительный вклад в случае МФЗ обычно неизвестны. Для консультирования необходимо использовать величины эмпирического риска, основанные на частоте повторных случаев заболевания во многих пораженных семьях. Эти величины обычно ниже величин риска для заболеваний, наследующихся по менделевским законам, и находятся в пределах 3-5% для многих частых врожденных пороков типа дефектов нервной трубки, заячьей губы и расщелины нёба. Для широко распространенных заболеваний среднего возраста, таких, как гипертензия, шизофрения и аффективные расстройства, риск для близких родственников (сибсов, родителей и детей) находится в пределах 10-15%. Следует всегда помнить о тщательном отграничении редкой моногенной формы заболевания, которое в общем случае наследуется мультифакториально. Так, некоторые больные подагрой являются носителями сцепленного с Х-хромосомой дефекта, обусловленного недостаточностью гипоксантин-гуанин - фосфорибозилтрансферазы (30800); причиной их подагры может быть и аутосомно-доминантная недостаточность фосфорибозилпирофосфат - синтетазы (13894). Среди мужчин в возрасте до 60 лет с ишемической болезнью сердца около 5% страдают семейной гиперхолестеринемией - аутосомно-доминантный признак (14440) (см. разд. 4.6.4).
Наследуемые аномалии хромосом, такие, как транслокации, часто не сегрегируют строго в соответствии с менделевскими соотношениями, и консультирование в этих случаях должно быть основано на величинах риска, полученных эмпирически (разд. 3.3.6; приложение 3).
Абсолютный риск (в %) является для семьи более важной величиной, чем информация об относительном риске, где частота заболевания сопоставлена с частотой в общей популяции. Для состояния, которое в популяции встречается с частотой 1:100000, стократное увеличение дает фактический риск, равный лишь 1:1000, т. е. риск повторного случая с практической точки зрения пренебрежимо мал. Для менделирующих заболеваний рекуррентный риск является постоянной величиной и не зависит от того, сколько пораженных родилось прежде-несколько или ни одного. У случая нет памяти! Напротив, для мультифакториальных заболеваний, таких, как врожденный порок сердца, расщелина губы или нёба, наличие в семье двух или более пораженных близких родственников означает, что в данной семье накоплено больше вредных генов и риск для будущих поколений становится выше обычных 3-5% [71]. Подробное обсуждение подходов к генетическому консультированию и оценке величин риска для многих типов заболеваний можно найти в недавно опубликованных книгах [71; 91].
Информация. О значении генетического риска больному нужно сообщать в форме, понятной для него. Следует информировать о том, что вероятность проявления серьезных пороков развития, наследственных заболеваний или умственной отсталости у детей нормальных родителей составляет 3-4% и эта величина является исходной мерой для оценки дополнительного риска. Могут возникнуть проблемы при сообщении о степени неопределенности. Например, при спорадическом случае порока развития, причины которого не установлены, риск может быть нулевым, если порок негенетического происхождения, и составлять 2-3% в случае мультифакториальной этиологии или даже 25%, если порок вызван аутосомно-рецессивной мутацией. Суммарный эмпирический риск, основанный на вероятности разных вариантов, часто представляется как эмпирический риск. В нашем примере такой риск может составлять 5% при допущении, что моногенные рецессивные разновидности этого порока развития встречаются редко. Однако многие консультируемые предпочитают, чтобы им говорили о неопределенности все, а не предлагали одну цифру риска [2265]. Необходимо четко объяснить тяготы, связанные с заболеванием. Очень тяжелые патологии со смертельным исходом в раннем детстве являются для семьи менее тяжким бременем, чем болезни, связанные с хронической инвалидностью. Нужно обсудить различные варианты при решении вопроса о целесообразности рождения ребенка. Поскольку обсуждаемая проблема может быть для консультируемого сложной и трудной в эмоциональном плане, иногда оказывается необходимым провести несколько бесед. В любом случае консультант должен дать письменное заключение, написанное понятным для непрофессионала языком.
Кровное родство. Двоюродные братья и сестры и более отдаленные родственники, собирающиеся вступить в брак, иногда обращаются за консультацией по поводу риска иметь детей с наследственными заболеваниями. Более чем в половине штатов США существуют законы, которые запрещают браки между двоюродными братьями и сестрами. Родство, несомненно, увеличивает риск заболевания, причиной которого служит гомозиготность по рецессивным генам (разд. 6.3), но абсолютный риск остается довольно низким. По приблизительным подсчетам вероятность различных заболеваний, пороков развития и случаев умственной отсталости среди потомства от браков двоюродных сестер и братьев превышает тот уровень, с которым сталкивается любая пара, не более чем вдвое. Таким образом, вероятность рождения в таком браке нормального ребенка будет около 93-95%. Для более отдаленных родственников величины риска еще ниже, и их трудно отличить от фоновой вероятности для такого рода расстройств. Какой-либо дополнительный риск для потомства нормального лица, состоящего в браке с неродственником, при условии, что родители одного из партнеров были кровными родственниками, отсутствует. С другой стороны, для детей от инцестных браков между братом и сестрой или отцом и дочерью (разд. 6.3.2.4), риск значителен. Вероятность того, что ребенок будет страдать тяжелой патологией, умственной отсталостью или умрет в детстве, составляет почти 50% (разд. 6.3.2.4). Поэтому рекомендуется, чтобы дети от инцестных браков, которых предполагается отдать на усыновление, наблюдались в течение примерно 6 месяцев до завершения процедуры усыновления. За это время большинство потенциальных дефектов должно проявиться. Заслуживает внимания тот факт, что дефекты, обнаруживаемые у потомства близкородственных браков, в большинстве случаев принимают форму неспецифических врожденных пороков развития, детской смерти и умственной отсталости, а не строго очерченных аутосомно-рецессивных заболеваний. Однако тщательный поиск различных врожденных ошибок метаболизма, связанных с рецессивными мутациями, в этих случаях не проводился, и вполне вероятно, что значительная часть детской смертности в инцестных браках обусловлена невыявленными врожденными ошибками метаболизма.
Возможно, что в обществах, где инбридинг практиковался в течение многих поколений (как в Индии, например), риск для потомства от близкородственных браков ниже, поскольку селекция против гомозиготных комбинаций генов должна была бы устранить многие такие гены через несколько поколений (разд. 6.3).
Выявление гетерозигот. Обнаружить гетерозиготность особенно важно у сестер мальчиков, страдающих сцепленными с Х-хромосомой заболеваниями типа гемофилии (30670) и миопатяи Дюшенна (30670). Независимо от генотипа мужа риск поражения тем же заболеванием сыновей гетерозиготных женщин составляет 50%. Аутосомно-рецессивные заболевания, напротив, проявляются в том случае, если оба родителя гетерозиготны; гетерозиготный брат или сестра больного должны вступить в брак с другим гетерозиготным лицом для того, чтобы у их детей заболевание проявилось. Вероятность того, что лицо, не являющееся близким родственником своего супруга - носителя аутосомно-рецессивного заболевания, тоже окажется носителем, обычно довольно низка.
Для выявления гетерозиготности могут быть полезны специальные лабораторные исследования, такие, как определение активности фермента креатинфосфокиназы при миопатии Дюшенна и определение свертывания крови с помощью антител к гемофилическому глобулину в сочетании с определением антигенной активности - при гемофилии А (30670) [2302; 2254].
Такие тесты, прежде чем применять их для идентификации отдельных носителей, требуют тщательной отработки и стандартизации на нормальных индивидах и облигатных гетерозиготах [2254]. Легко и просто гетерозиготность выявляется при гемоглобинопатиях. Появляется возможность идентифицировать носительствО по большому числу различных аутосомных ферментативных дефектов, таких, например, как недостаточность гексозаминидазы при болезни Тея - Сакса (ранняя детская амавротическая идиотия) [2320]. Если данные лабораторных испытаний перекрываются в области низких значений для нормы и высоких значений для носителей, то смысл одинаковых данных у разных индивидов можно трактовать по-разному в зависимости от априорной вероятности носительства у тестируемого лица. Тесты, которые прекрасно служат для обнаружения носительства у сестер мужчин, страдающих Х-сцепленными заболеваниями, могут давать слишком много "ложноположительных" ответов при обширных скрининговых исследованиях родственников и особенно населения в целом [2300; 2349]. Например, при использовании тех же стандартов, которые с высокой вероятностью обнаруживают гетерозиготность у сестер мальчиков, страдающих гемофилией, 5% женщин нормальной популяции были бы идентифицированы как носители гемофилии. В табл. 9.3 перечислены болезни, для которых возможно и необходимо определять носительство.

Таблица 9.3. Наследственные болезни: выявление гетерозиготного носительства*, при решении вопроса о целесообразности рождения детей
* (Ограничено транслокационным синдромом Дауна, Х-сцепленными и распространенными аутосомно-рецессивными заболеваниями. Определение гетерозиготности по генам, обусловливающим недостаточность ферментов, возможно, но для нормальных сибсов больного риск иметь пораженное потомство очень мал, поскольку для таких врожденных ошибок метаболизма частота состояния носительства в популяции чрезвычайно низкая.)
В некоторых ситуациях для уточнения генетического прогноза могут оказаться полезными дополнительные статистические методы. Например, брат женщины и ее дядя по материнской линии страдают сцепленным с Х-хромосомой заболеванием. Она, следовательно, имеет 50%-ный риск быть гетерозиготой. Предположим, что у нее уже есть два здоровых сына и что тест для обнаружения гетерозиготности выполнить невозможно. Сведения о том, что два ее сына нормальны, уменьшают вероятность того, что она является носителем. С другой стороны, тест, который выявляет 90% гетерозигот, может дать для этой женщины отрицательный результат. В этом случае ее риск оказаться носителем очень низок. В приложении 8 и в книге Мерфи и Чейза [149] даются статистические принципы расчета точных величин риска повторных случаев в таких ситуациях.
Увеличивающееся разнообразие и доступность ДНК-маркеров дают возможность более эффективно диагностировать носительство для сцепленных с Х-хромосомой заболеваний (табл. 9.2). Уже существует несколько зондов (как Х-хромосом специфичных, так и ген-специфичных), которые распознают варианты ДНК, связанные с генами гемофилии А и Б. Если использовать несколько зондов для гемофилии А, все женщины оказываются гетерозиготными по тому или другому из этих вариантов. Поэтому диагноз носительства, как правило, можно устанавливать в тех семьях, где ген гемофилии А сегрегирует. Для миопатии Дюшенна ситуация аналогичная: здесь также имеются ДНК-маркеры, тесно сцепленные с геном миопатии. Информацию, полученную с помощью ДНК-маркеров, можно дополнить данными тестирования на креатинкиназу и сведениями о родственниках (см. приложение 8 для примера подробных расчетов). В настоящее время ведется интенсивная работа, цель которой получить ДНК-зонды для Х-сцепленной умственной отсталости, что даст возможность выявлять носителей и этой патологии.
Принятие решения и альтернативы. Если супруги решают, что риск иметь больного ребенка слишком велик, нужно обсудить с ними несколько вариантов кроме контрацепции. Усыновление практикуется все реже из-за небольшого числа подходящих для такого шага детей. Можно предложить кому-то из супругов стерилизацию, но необходимо подчеркнуть, что эта процедура имеет необратимый характер и потому нежелательна. Ведь после возможного развода или смерти одного из супругов повторный брак может практически полностью устранить риск, если партнер по второму браку не является носителем. Еще одна возможность, которую следует обсудить, - это искусственное оплодотворение. Понятно, что этот вариант подходит только для тех случаев, когда опасность аутосомно-рецессивного или аутосомно-доминантного заболевания исходит от мужа.
Выявление генетических заболеваний у родственников. Оптимальное генетическое консультирование при некоторых заболеваниях должно включать проверку родственников с повышенным риском (табл. 9.4). При некоторых состояниях выявление латентной формы заболевания у родственников может спасти жизнь, если сопровождается соответствующим лечением. Близкий родственник пациента с болезнью Вильсона имеет 25%-ную вероятность развития того же заболевания, но может быть слишком молод для проявления клинических симптомов. Сибсы больного наследственным полипозом (17510) [2391] имеют 50%-ную вероятность заболевания и определенный риск злокачественной трансформации одного или нескольких полипов при этом состоянии. В общем, нужно принимать решительные меры для обследования родственников, если генетическое состояние связано с серьезными заболеваниями, которые можно предотвратить или лечить. Например, обследование родственников совершенно необходимо при заболевании типа поликистоза почек (17390) [2276; 2369а]. Раннее выявление этой патологии у кого-то из них дает возможность разумно выбрать профессию, определить подходящий образ жизни, решить вопрос о целесообразности рождения детей и, наконец, привыкнуть к мысли о необходимости в будущем пересадки почки или диализа. Нужно искать в семьях возможных носителей серьезных заболеваний, сцепленных с Х-хромосомой (гемофилия, миопатия Дюшенна, например), и хромосомного статуса (типа синдрома Дауна, связанного с транслокацией) для предотвращения с помощью пренатальной диагностики (см. ниже).
Таблица 9.4. Наследуемые по аутосомно-доминантному типу генетические заболевания зрелого возраста, поддающиеся лечению и предупреждению, для которых обязателен поиск среди членов семьи больного
Директивное и недирективное генетическое консультирование. После проведения генетической консультации, которая включает оценку рекуррентного риска (см. ниже), родителям нужно решить, иметь им еще детей или нет. Многие врачи склонны давать директивные советы в отношении будущей беременности (за или против). Напротив, в практике медицинской генетики довольно сильна тенденция не давать определенных указаний. Отчасти это связано с социологическими причинами. В Соединенных Штатах Америки, где генетическое консультирование впервые появилось около 30 лет тому назад, оно обычно проводилось генетиками, не имевшими профессиональной медицинской подготовки и привычки к предписаниям. Подобного рода недирективность генетического консультирования хорошо соответствует возникшей недавно тенденции предоставить большую самостоятельность пациенту в принятии решения. Поскольку каждая семья единственна в своем роде и отличается по реакции на риск, недирективность благоприятствует принятию зрелого решения. Однако абсолютно нейтральное консультирование вряд ли возможно, да и нежелательно. Любой человек (или семья), который обращается за консультацией, хочет большего и нуждается в большем, нежели в похожем на компьютер профессионале, выдающем только факты. Консультирующий может сделать акцент на более обнадеживающих или, наоборот, на более негативных аспектах данного заболевания. Эти настроения прямо или косвенно будут влиять на ход мыслей в процессе генетического консультирования. Чаша может быть наполовину полной или наполовину пустой - все зависит от отношения: можно более решительно подчеркивать либо позитивные, либо негативные аспекты ситуации. Далеко не все пары имеют уровень образования, достаточный, чтобы принять обдуманное решение. Они рассчитывают, что специалист по медицинской генетике, имеющий необходимый опыт и знания, поможет им принять решение. "Что бы Вы сделали на моем месте?" - такой вопрос часто задают консультируемые независимо от их биографических данных. Однако в силу того, что жизненные обстоятельства, религиозные и культурные традиции супружеских пар и консультанта могут быть совершенно разными, предпочитаемый консультантом выбор не обязательно адекватен для супругов. Пары часто принимают разные решения относительно рождения ребенка, даже если генетические данные и суть заболевания идентичны. При некоторых заболеваниях, например при хорее Гентингтона, прогноз такой мрачный, что многие консультанты занимают весьма твердую позицию и убеждают тех, чей риск составляет 50%, воздерживаться от потомства. Кроме того, разные страны отличаются по своим культурным традициям. По нашим представлениям, директивное консультирование более характерно для Центральной Европы и социалистических стран, нежели для англоязычного мира.
Оценка генетического консультирования и психологические аспекты [2289; 2332; 2382]. Генетическое консультирование является относительно новым направлением, и его практика еще не устоялась окончательно. Большинство специалистов в этой области согласны с тем, что консультируемые должны хорошо понять медицинское и социальное значение заболевания, чтобы иметь возможность принять решение о целесообразности рождения детей. Некоторые исследователи определяли эффективность генетического консультирования путем оценки последующего поведения супругов. Генетическое консультирование считалось успешным, если число пар, воздерживающихся от рождения детей, было большим среди пар с высоким (более 10%) риском, чем среди пар с низким. В нескольких исследованиях действительно отмечен такой результат [2271]. Однако столь узкую задачу нельзя считать истинной целью генетического консультирования. Было бы лучше выяснить, достигнуто ли полное понимание природы болезни и все ли требования, касающиеся информации, психологической и социальной поддержки, были удовлетворены. Различные исследования, посвященные генетическому консультированию, сходятся в том, что и после консультирования многие пациенты толком не понимают сути заболевания и путаются в понятии повторного риска. Самое большое исследование, которое охватывало 205 консультантов и свыше 1000 консультируемых женщин, было проведено группой социологов в конце 70-х гг. на базе 47 клиник по генетическому консультированию в США [2382]. Исследование включало много разных генетических состояний и консультантов. Пациентов просили дать оценку процессу консультирования и рассказать о своих переживаниях. Результаты показали, что во время консультации врачи-генетики склонны делать акцент на вероятности повторных случаев в семье, тогда как консультируемых больше интересуют причины, прогноз и лечение заболевания - вопросы, которые, по их мнению, часто не обсуждаются в той мере, в какой хотелось бы. В процессе консультации речь идет о генетических аспектах болезни, между тем пациентов обычно беспокоят психосоциальные проблемы, которыми медицинская генетика не занимается.
Это исследование показало, что 54% консультируемых, которым сообщили о риске, и 40% консультируемых, которым поставили диагноз, были неспособны рассказать об этом вскоре после консультации. Это обстоятельство не было связано с тем, кто конкретно проводил консультацию - доктор медицины или генетик-ассистент, и не зависело от опыта консультанта. У консультантов с многолетним опытом работы результаты были не лучше, чем у недавних выпускников. В нескольких других исследованиях результаты в плане понимания риска были существенно лучшими, но никоим образом не совершенными [2289]. Как правило, но не всегда, уровень понимания положительно коррелировал с образованием пациента.
Семьи с высоким уровнем образования более интенсивно используют службы генетического консультирования, чем группы населения, имеющие более низкий образовательный ценз. Пары, которые заинтересованы в том, чтобы разобраться в природе заболевания и оценить повторный риск, с большей вероятностью окажутся при выборе решения под влиянием полученной информации, чем те, которых на консультацию прислали и которые так и не поняли цель этого мероприятия. Таким образом, пациенты, обращающиеся за генетической помощью самостоятельно, как правило, лучше понимают сообщаемую им информацию. В другом исследовании изучали характер восприятия информации при консультировании [2334; 2335]. Оказалось, что пациенты часто не воспринимают степень риска, представляемую цифрами, в вероятностном смысле. Их одолевает множество сомнений, касающихся того, как сделать правильный выбор, как окружающие отнесутся к их решению, что значит иметь больного ребенка и смогут ли они хорошо исполнить по отношению к нему свои родительские обязанности. Эти данные показывают, что ход мыслей консультанта и консультируемых часто не совпадает: консультируемым оказывается трудно иметь дело с вероятностной информацией. Перекинуть мост через эту пропасть - проблема нелегкая.
Генетическое консультирование в том виде, в котором оно практикуется в настоящее время в большинстве стран, меньше уделяет внимания эмоциональным аспектам по сравнению с консультированием, осуществляемым в других областях, например в области психологических и брачно-семейных отношений. Некоторые исследователи полагают, что необходимо обращать больше внимания именно на психодинамические аспекты наследственной патологии [2325; 2270; 2287]. Наш собственный опыт, однако, говорит о том, что психологически ориентированное генетическое консультирование, при котором значительное время уделяется психологии и психодинамике, требуется нечасто. Лучшим выходом из ситуации, когда имеются серьезные психологические проблемы, является направление на консультацию к психиатру или психотерапевту.
Хотя генетику-консультанту и нет необходимости проводить развернутое психологическое консультирование (оно потребовало бы более интенсивной стажировки в клинической психологии, чем дается в настоящее время), он должен помнить, что его роль не сводится к равнодушной выдаче информации.

Таблица 9.5. Пренатальный диагноз наследственных болезней и пороков развития
Пренатальная диагностика [2312; 2357; 2373; 2286]. В последние годы область пренатальной диагностики бурно развивалась, что во многом изменило практику генетического консультирования. В ходе беседы консультант теперь сообщает о возможности дородовой диагностики. Вполне понятно, что прямой ответ на вопрос о здоровье будущего ребенка гораздо более ценен, чем сведения о вероятности повторного случая. Дородовая диагностика включает ряд методов, среди которых чаще всего используются амниоцентез (пункция околоплодного пузыря) и ультразвуковое обследование.
Пункция околоплодного пузыря (рис. 9.2). Пункция проводится в начале второго триместра беременности (15-17 нед) через брюшину. Эта процедура оказалась безопасной в руках опытных акушеров, но она безвредна не на 100%. Есть небольшой риск потери плода (0,5%). Инфекция и гематома случаются намного реже; еще более редки другие осложнения. Процедура проводится в амбулаторных условиях в сочетании с ультразвуковым обследованием, которое уменьшает долю неудач и возможность появления окрашенной кровью жидкости и кровоизлияния у плода и матери. Обычно проводится исследование хромосом, которое требует культивирования отсосанных из пузыря клеток, принадлежащих плоду, поэтому результаты получают через 2-3 недели. Кроме хромосомных аберраций в амниотических клетках можно обнаружить дефекты многих ферментных систем (табл. 9.6). Поскольку недостаточность некоторых ферментов встречается редко, а биохимический анализ сопряжен со значительными методическими трудностями, соответствующее исследование в идеале должно проводиться специализированными лабораториями, куда отсылается материал. Биохимическое исследование (в отличие от поиска аберрантных хромосом у пожилых матерей) в целях пренатальной диагностики проводится не в обязательном порядке, а только по специальным показаниям при беременности, сопряженной с высоким риском (т. е. при наличии уже одного больного ребенка).
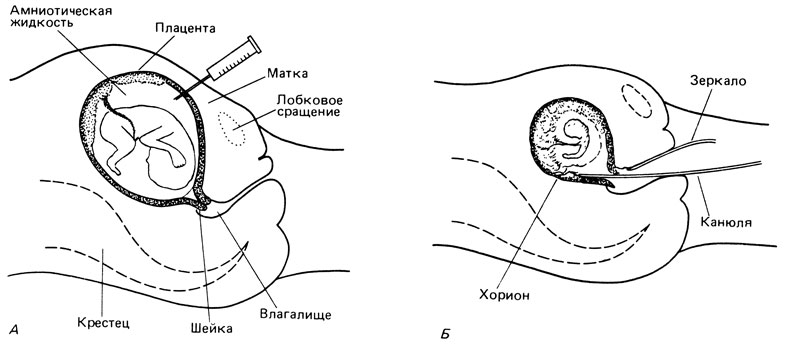
Рис. 9.2. А. Амниоцентез. Пункция околоплодного пузыря через брюшную стенку. Б. Взятие пробы эпителия ворсинок хориона. В матку проникают через влагалище и шейку матки
![Таблица 9.6. Пренатальная диагностика врожденных наследственных ошибок метаболизма [308]](pic/000083.jpg)
Таблица 9.6. Пренатальная диагностика врожденных наследственных ошибок метаболизма [308]
Исследование ворсинок хориона [2267; 2318; 2323; 2346; 2379; 2399]. В течение последних нескольких лет в практику пренатальной диагностики было введено взятие пробы ворсинок эпителия хориона. Эту процедуру проводят под ультразвуковым контролем и получают ткань хориона (происходящую из трофобласта плода) для цитогенетического, биохимического исследований и анализа ДНК. Процедуру можно проводить между 8-й и 10-й неделями беременности, поэтому для пациентки она имеет психологические преимущества перед амниоцентезом, который проводится на 15-17-й неделе беременности. Результаты цитогенетического исследования можно получить сразу или в течение одного дня (благодаря наличию делящихся клеток). Риск, связанный с этой процедурой, до конца еще не выяснен, и в настоящее время проводятся интенсивные клинические исследования, направленные на его оценку. Особенно тщательно исследуется проблема риска абортов и диагностических ошибок, обусловленных мозаицизмом по хромосомным аберрациям.
Ультразвуковая эхография [2268]. Широкое использование неинвазивного ультразвукового обследования плода позволяет в настоящее время проводить пренатальную диагностику многих аномалий развития. В табл. 9.7 перечислены различные заболевания, которые можно диагностировать этим способом. Существенное улучшение оборудования для эхографии, которое имеет место в последние годы, дает возможность лучше различать детали морфологии плода. Однако в диагностике многое зависит от знаний и опыта операторов, проводящих ультразвуковое обследование.

Таблица 9.7. Ультразвук в пренатальной диагностике
Все современные исследования указывают на то, что ультразвук не вредит развивающемуся плоду, однако применение этого метода без разбора вызывает некоторое беспокойство: ведь абсолютных доказательств его безвредности не получено. Различные авторитетные учреждения (Национальный институт здоровья, США, Всемирная организация здравоохранения) советуют соблюдать осторожность и использовать ультразвуковую эхографию только при наличии определенных показаний со стороны матери или плода. Между тем в некоторых странах повторное ультразвуковое обследование стало частью рутинного наблюдения за ходом беременности. Надо признать, что это позволило диагностировать многие пороки развития, о существовании которых ранее не подозревали.
Эндоскопия плода [2342; 2374]. Эндоскопия плода обычно проводится между 18-й и 22-й неделями беременности с помощью миниатюрных волоконно-оптических инструментов, вводимых в полость амниона. Проводимая даже опытными руками эта процедура в 5-10% случаев влечет за собой выкидыш. Из-за узкого поля зрения обследование плода с целью выявления дефектов развития ограничено. Под прямым визуальным контролем можно взять пробу крови плода, и это используется для диагноза талассемий, а также гемофилии и синдрома фрагильной Х-хромосомы. Можно диагностировать любой генетический дефект, который проявляется в клетках крови. Можно взять биопсию кожи плода или даже биопсию печени, что применялось для диагностики заболеваний, которые проявляются только в патологии печени.
Взятие крови у плода. Для получения клеток крови плода с целью анализа на гемоглобинопатию раньше применяли пункцию плаценты, однако из-за относительно высокой доли кровоизлияний у плода и необходимости повторять процедуру такое исследование теперь проводится довольно редко.
Взятие проб крови матери [216; 2274; 2298; 2342]. В качестве скринингового метода для выявления дефектов нервной трубки и некоторых других аномалий плода (табл. 9.8) во многих центрах проводилось исследование проб крови матери (взятых с помощью венопункции) на повышенное содержание альфа-фетопротеина. Поскольку дефекты нервной трубки в большинстве стран встречаются все реже, а возможность их выявления с помощью ультразвуковой эхографии растет, трудно с определенностью говорить о целесообразности повсеместного применения такого скрининга. Скрининг альфа-фетопротеина может оказаться полезным при выявлении синдрома Дауна, поскольку плоды с трисомией по 21-й хромосоме имеют уровень альфа-фетопротеина более низкий, чем нормальные (примерно 0,7 от нормального) [2261; 2275]. Было показано, что если бы амниоцентез и хромосомное исследование проводились всем женщинам с уровнем альфа-фетопротеина в крови выше определенной величины (скажем, 0,5 от среднего уровня), то выявилось бы 20-40% случаев синдрома Дауна в дополнение к тем, которые обнаруживаются современными методами, использующими амниоцентез у пожилых беременных женщин. Однако на каждый случай выявляемой таким способом трисомии по 21-й хромосоме нужно было бы провести 150-200 дополнительных амниоцентезов при нормальной беременности.

Таблица 9.8. Нарушения, которые увеличивают и уменьшают уровень альфа-фетопротеина в околоплодной жидкости
В процессе исследования находятся и другие методы, например выявление клеток плода в системе кровообращения матери. Успешное применение такого метода было бы полезным при скрининге материнской крови на содержание клеток плода с хромосомными и биохимическими аномалиями. Однако, даже если клетки плода присутствуют в крови матери, предстоит преодолеть многие технические трудности, прежде чем эту процедуру можно будет применять в обычном порядке.
Показания для пренаталъной диагностики. Для относительно новых видов пренаталъной диагностики существует больше показаний, с ее помощью можно диагностировать все большее число дефектов. Исследование ворсинок эпителия хориона проводится по тем же показаниям, что и амниоцентез, но имеет то преимущество, что может проводиться на существенно более ранних сроках беременности.
1. Возраст матери. Амниоцентез чаще всего проводят для того, чтобы исключить синдром Дауна и другие хромосомные аберрации у женщин "пожилого" для материнства возраста. В большинстве стран этот возраст несколько произвольно определен в 35 лет. Риск для синдрома Дауна в этом возрасте составляет около 1/400, к 40 годам он возрастает до 1/100 и к 44 годам - до 1/40 (табл. 5.4). При амниоцентезе синдром Дауна и другие хромосомные аберрации встречаются значительно чаще, чем при родах, так как многие анеусомики абортируются до рождения (табл. 5.4).
2. Предшествующий анеусомик. Предыдущий ребенок с синдромом Дауна или трисомией по другой хромосоме несколько увеличивает риск повторного случая. Для синдрома Дауна риск составляет в этом случае около 1/250 для матерей в возрасте менее 35 лет, а для тех, кому больше 35 лет, примерно вдвое превышает риск, специфичный для возраста.
3. Перестройки родительских хромосом [310; 334]. Носительство транслокаций или перицентромерных инверсий увеличивает риск несбалансированной транслокации и аномального плода (т. е. транслокационного синдрома Дауна) (разд. 2.2.2). Риск не соответствует величинам, ожидаемым по сегрегации хромосом, возможно из-за селекции, направленной против несбалансированных гамет, и базируется на эмпирических данных. Риск для транслокационной трисомии 21 составляет около 15%, если носителем является мать, и только 3%, если носителем является отец (t14q21 и t21q22q).
При реципрокных транслокациях риск поражения будущего потомства существенно выше (около 20%), если он определяется через живых пораженных потомков, а не по рецидивам выкидышей (5%-ный риск) (подробно об этом см. разд. 2.2.2.2). Более обширные несбалансированные дупликации/делеции (3-6 хромосомных сегментов из общего их числа около 200) связаны с меньшим риском рецидива (9-16%), чем транслокации с дупликациями/делециями, затрагивающими только 1-2 сегмента (34%). Возможно, эмбрионы с более крупными дефектами часто нежизнеспособны и спонтанно абортируются еще до проведения амниоцентеза.
4. Риск для дефектов, сцепленных с Х-хромосомой. В том случае, если плод мужского пола у данной матери имеет 50%-ный риск серьезного Х-сцепленного заболевания, проводят определение пола эмбриона. В этой ситуации для большинства родителей биопсия ворсинок хориона на ранних сроках беременности более приемлема, чем амниоцентез. Усовершенствование методов анализа ДНК даст возможность не использовать пол плода в качестве диагностического критерия.
5. Синдром фрагилъной Х-хромосомы. Диагностика этого синдрома общей умственной отсталости (разд. 8.2.1.2) требует взятия пробы крови плода при эндоскопии, поскольку с помощью амниотических клеток этот дефект пока еще нельзя диагностировать надежно.
6. Гемоглобинопатии [2345]. До недавнего времени для выявления различных талассемий анализировали клетки крови плода, полученные с помощью эндоскопии. В настоящее время этот метод замещается ДНК-диагностикой с использованием клеток плода, получаемых при биопсии ворсинок эпителия хориона. Для серповидноклеточной анемии возможно прямое выявление мутации (см. разд. 4.3), тогда как для диагностики талассемий выявляют сцепление с различными ДНК-маркерами.
7. Врожденные ошибки метаболизма. Чтобы обнаружить врожденные ошибки метаболизма, необходимо провести анализ ферментных систем клеток плода. Перечень таких состояний приведен в табл. 9.6. При классической фенилкетонурии, при которой фермент не выявляется в амниотических клетках, возможна диагностика с помощью метода гибридизации ДНК (метод сцепления с ДНК-маркерами).
8. Различные наследственные заболевания, диагностируемые методом сцепления с ДНК-маркерами. Перечень этих заболеваний приведен в табл. 9.2, а обсуждение - в разд. 2.3. Этот список растет.
9. Дефекты нервной трубки. У женщин с высоким риском, т. е. уже имеющих больных детей или обнаруживающих высокий уровень альфа-фетопротеина в крови, следует проводить амниоцентез (но не биопсию ворсинок хориона) для определения уровня альфа-фетопротеина в амниотической жидкости. Кроме того, в этом случае очень полезно ультразвуковое обследование. Пренатальная диагностика довольно широко используется в развитых странах, хотя во многих регионах значительная доля беременных женщин в возрасте свыше 35 лет не обследуется. В Дании широкая информация общественности через женские журналы была воспринята наилучшим образом: 80% пожилых беременных проходят обследование. Доступность пренатальной диагностики часто побуждает родителей, которые воздерживались от рождения детей из-за страха иметь больного ребенка, сделать такую попытку. Хотя во многих странах стали благосклонно относиться к искусственному прерыванию беременности из-за выявленного у плода дефекта, значительная доля населения Соединенных Штатов и других стран по религиозным и другим причинам настроена против аборта в этих случаях. Они считают, что такая практика является началом "скольжения по наклонной плоскости", которое неминуемо приведет к стремлению отвергнуть относительно небольшие дефекты в поиске "совершенного" ребенка и возрождению евгенических и расистских проектов. Высказываются опасения, что общество не захочет заботиться о детях с наследственными заболеваниями в ситуации, когда аборт мог бы предотвратить их рождение. Однако, поскольку в настоящее время большинство генетических дефектов предотвратить не в наших силах, такую опасность нельзя считать реальной. Более того, против этих опасений говорит тот факт, что в большинстве стран в последние годы финансовая и социальная помощь страдающим этими недугами людям увеличилась.
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'