Приложение 8
Медико-генетическое консультирование: использование условных вероятностей
Проблема оценки генетического риска. Как отмечалось в разд. 9.2.1, получение оценки генетического риска основывается либо на сегрегационных отношениях в случае менделирующих заболеваний, либо на цифрах эмпирического риска, если тип наследования сложен. При отсутствии дополнительной информации такие цифры прямо используются для расчета конкретного риска для определенного пробанда или семьи. Например, каждый будущий ребенок пораженного члена большой родословной, собранной Фараби (разд. 3.2.1, рис. 3.2), будет обладать 50%-ным риском иметь брахидактилию. Однако существует немало ситуаций, в которых для получения оценки риска может оказаться полезной дополнительная информация.
Пример: наследственная и спорадическая ретинобластома. Как отмечалось в разд. 5.1.6, ретинобластома, врожденная глазная опухоль у маленьких детей, проявляется либо как доминантное заболевание примерно с 90%-ной пенетрантностью, либо как спорадический случай, вероятно, вследствие соматической мутации. В последнем случае оба родителя (и все другие члены семьи) не поражены, и генетический риск для ребенка не выше, чем частота в общей популяции, т. е. около 1:15000 - 1:25000. Кроме того, соматическая мутация всегда приводит к односторонней ретинобластоме. Однако среди всех спорадических случаев примерно 10% вызываются мутацией, возникшей в гаметах одного из родителей. В таких случаях родители и другие члены семьи также не поражены, но для каждого ребенка пробанда со спорадической односторонней ретинобластомой риск иметь опухоль составляет уже около 45% (90%-ная пенетрантность при 50%-ном сегрегационном отношении). Рассмотрим следующую ситуацию: больной со спорадическим односторонним поражением спрашивает о генетическом риске для своих детей. Если нет другой информации, то риск составляет 0,9 × 0% (для спорадических соматических случаев) + 0,1 × 45% (для спорадических гаметических случаев) = 4,5%. Однако ситуация становится сложнее, если пробанд уже имеет по крайней мере одного здорового ребенка. Если бы его болезнь была вызвана доминантной мутацией, то каждый из его детей имел бы 45%-ный риск быть пораженным. Наличие по крайней мере одного здорового ребенка увеличивает вероятность, что у пробанда на самом деле имеется ненаследственная форма болезни, а это снижает риск для его будущих детей. Как вычислить этот риск?
Вероятность иметь наследственную форму болезни [2393]. Как уже отмечалось, априорная вероятность того, что наш пробанд имеет наследственную форму, равна Р(Н) = 0,1. Если он имеет наследственную форму, то условная вероятность, что его первый ребенок окажется непораженным (событие 17), т. е. вероятность, что он не поражен, несмотря на тот факт, что пробанд несет этот ген, равна P(U/H) = 0,55. С другой стороны, априорная вероятность того, что пробанд имеет ненаследственную форму, равна Р (не Н) = 0,9. В этом случае условная вероятность того, что его ребенок окажется непораженным, будет Р(U/не Н) - 1, потому что риска (почти) нет. Отсюда можно получить формулу для его апостериорной вероятности иметь наследственную форму
| P(H/U) = | P(H)×P(U/H) | . (П.8.1) |
| P(H)×P(H/U) + P(не H)×P(U/ не H) |
Подстановка значений из нашего примера дает
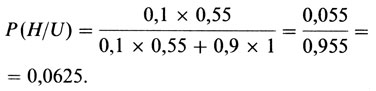
Следовательно, один непораженный ребенок снизил нашему пробанду вероятность иметь наследственную форму с 0,1 до 0,058. Риск быть пораженным для его следующего ребенка равен теперь
или снизился с 4 до 2,6%. При двух непораженных детях условная вероятность P(U/H) становится 0,552 = 0,3025. Подстановка в уравнение П.8.1 приводит к P(H/U) = 0,0325, R2 = 0,015. Для n детей P(U/H) становится равной 0,55n. Суть этого принципа легко уловить из рис. П.8.1.
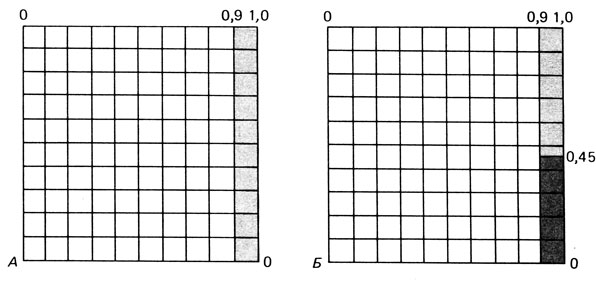
Рис. П.8.1. Графическое представление процедуры расчета риска для детей, родители которых страдают односторонней ретинобластомой. А. Светло-серая область представляет родителей с наследственной ретинобластомой (примерно 10% всех односторонних спорадических случаев). Б. После рождения первого ребенка 45% этих родителей (= 4,5% всех родителей) оказываются носителями наследственной формы. Только 5,5/95,5% остатка (что соответствует нормальному первому ребенку) будут носителями генов наследственной формы
Удобная система обозначений и форма графического представления. Мерфи [149] предложил ясную и удобную систему записи, которая делает описанное выше вычисление более очевидным, особенно для медиков-профессионалов, которые, как правило, испытывают трудности при оперировании абстрактными математическими понятиями. Конструируется таблица, в которой визуализуется пошаговое вычисление. В табл. П.8.1 приведено описанное выше вычисление для ретинобластомы. Исходя из апостериорной вероятности для нашего пробанда иметь наследственную форму (0,58), вероятность того, что первый ребенок будет поражен, можно вычислить так, как было показано в предыдущем пункте:
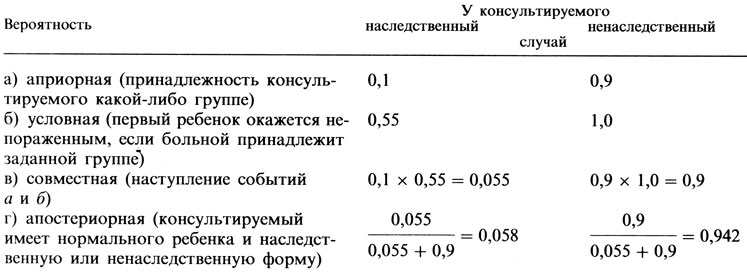
Таблица П.8.1. Вычисление вероятности для ретинобластомы
Принцип вычисления можно представить также графически (рис. П.8.1). В изображенном на рисунке квадрате неокрашенная область представляет группу ненаследственных случаев, а слабо окрашенная область - наследственные случаи. После того как первый ребенок пережил опасный период (первые несколько лет жизни), родители пораженного ребенка (а именно 45% наследственных случаев, рис. П.8.1, темная область) исключаются из группы больных спорадической односторонней ретинобластомой. Эти 45% четко установлены как наследственные с соответствующим 45%-ным риском для последующих детей. Риск для пробандов со здоровыми детьми следует вычислять на основе общей области, за исключением темной части. Теперь наследственные случаи представлены не более чем 10/100 квадратов (5,5/95,5).
Пример: хорея Гентингтона. Здоровый мужчина в возрасте 35 лет обратился в медико-генетическую консультацию. Его отец и бабка страдают хореей Гентингтона. Он интересуется риском заболеть для себя и своих будущих детей. Хорея Гентингтона - это аутосомно-доминантное заболевание с полной пенетрантностью. Возраст начала варьирует от 20 до 70 лет (разд. 3.1.2, рис. 3.4). Если бы у пробанда были обнаружены признаки болезни, то проблема была бы простой: каждый из его детей имел бы 50%-ный риск. Если на самом деле он еще не достиг возраста манифестации, то проблема также была бы простой: он имел бы 50%-ный риск, а его дети - 50% от 50%, т. е. 25%-ный. Однако реально он уже прожил часть периода манифестации и остался здоровым. Этот факт увеличивает его шанс быть гомозиготой по нормальному аллелю и оставаться непораженным. Как эта ситуация влияет на риск для его детей? В возрасте 35 лет примерно 30% всех гетерозигот уже обнаружили клинические признаки болезни. Это ведет к следующим расчетам:

Такие расчеты можно провести для многих других конкретных ситуаций при аутосомно-доминантных и рецессивных заболеваниях (детальное обсуждение см. в [71]).
Гетерозиготы по Х-сцепленным рецессивным заболеваниям. Этот тип вычислений наиболее важен при консультировании женщин, для которых существует риск оказаться гетерозиготами по Х-хромосомному рецессивному признаку и иметь пораженных сыновей. Обратимся к родословной на рис. П.8.2. В сущности Альма определенно является гетерозиготой, поэтому ее дочь Барбара имеет априорную вероятность 50% также быть гетерозиготой. Это означает 0,5 × 0,5 = 0,25 - риск для любого ее сына проявить признак. Если нет другой информации, то приведенные выше значения формируют основу для консультирования.
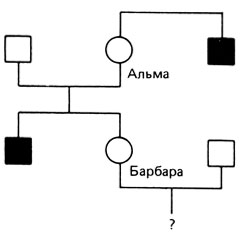
Рис. П.8.2
Барбара является носителем: 0,5 Риск для сына: 0,5 × 0,5 = 0;25
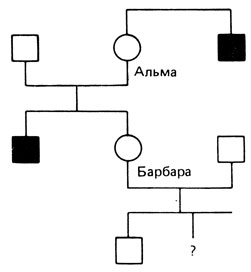
Рис. П.8.3
В родословной на рис. П.8.3 ситуация иная. В этом случае у Барбары уже имеется нормальный сын. Условная вероятность иметь нормального сына для гетерозиготной Барбары равна 0,5. Вычисления выполняются следующим образом:

Вычисление производится точно так же, если родословная сложнее, т. е. если у Барбары есть дочь и она захотела узнать риск для своих сыновей и т. д. В этом случае апостериорную вероятность Барбары нужно использовать для получения априорной вероятности ее дочери. Ряд конкретных примеров можно найти в [71].
Ситуация оказывается существенно иной, когда случай болезни рассматривается как спорадический (рис. П.8.4). Больной может быть либо носителем новой мутации, когда его мать-нормальная гомозигота, и риск для сыновей ее сестер не увеличивается, либо мать гетерозиготна и ее сестры также имеют априорную вероятность 0,5 быть гетерозиготами. Как упоминалось в разд. 5.1.3.4, доля новых мутантов среди носителей (редкого) Х-сцепленного рецессивного признака равна
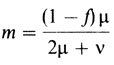
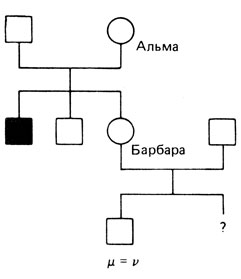
Рис. П.8.4
(f - относительная плодовитость носителей признака по сравнению с общей популяцией, μ - уровень мутаций в женских клетках, v - уровень мутаций в мужских гаметах). Когда уровни мутаций равны у двух полов f = 0, то m = 1/3. Это означает, что мать имеет априорную вероятность 2/3 быть гетерозиготой. Это ведет к следующему расчету риска для сына Барбары:

Такое простое вычисление справедливо только, если выполняются упомянутые выше два условия (μ = v, f = 0) и существует генетическое равновесие между мутационным процессом и отбором. Так бывает при мышечной дистрофии Дюшенна - наиболее распространенной Х-сцепленной рецессивной болезни во многих популяциях. Для других заболеваний, таких, как гемофилия А и недостаточность гипоксантинфосфорибозил-трансферазы, частоты мутаций оказываются намного выше в мужских гаметах, чем в женских (разд. 5.1.3.4). Здесь долю т нужно вычислять на основе эмпирических данных. Приемлемой аппроксимацией будет v = 10 × μ, поскольку уровень мутаций в мужских гаметах примерно в 10 раз выше, чем в женских. При отсутствии каких-либо определенных данных можно принять априорную вероятность 1 для Альмы и 1/2 для Барбары (рис. П.8.4), что немного завышает риск.
Ниже мы приведем более сложный пример, другие аналогичные можно найти в [149; 71; 205]. Родословная изображена на рис. П.8.5. У Барбары два брата: один поражен, а другой - нет. Кроме того, у нее имеется сестра Беттина, у которой двое здоровых сыновей. Беттина является либо нормальной гомозиготой (1/2, если Альма гетерозиготна), и в этом случае следует ожидать нормальных сыновей, либо она гетерозиготна, и в этом случае условная вероятность иметь двух нормальных детей составляет 1/4. Эта оценка (1/2 + 1/4 × 1/2 = 5/8) включается в вычисление условной вероятности для Альмы, причем учитывается также ее условная вероятность иметь непораженного сына, если она гетерозиготна. Вычисление проводится следующим образом (снова μ = v, f = 0):

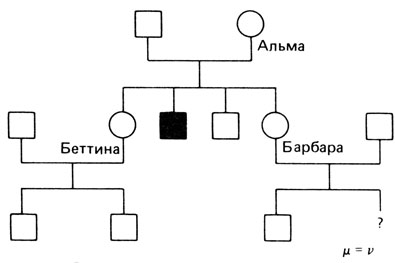
Рис. П.8.5
Все приведенные расчеты рисков (быть носителем) основывались только на информации о родословной. На практике для уточнения риска следует использовать дополнительные данные, которые можно получить из биохимических и молекулярных исследований. При мышечной дистрофии Дюшенна иногда повышается уровень креатинкиназы у гетерозигот, но однозначно дифференцировать гетерозигот и нормальных гомозигот на основе соответствующих значений невозможно (обсуждение тестов на гетерозиготность при этом заболевании можно найти в разд. 4.2.2.8). Подобный подход применяется при диагностике носительства по гемофилии. В этом случае используются иммунологический тест и анализ на присутствие фактора свертывания крови VIII.
В идеале в каждом регионе должна существовать одна лаборатория, которая занимается проведением анализов, направленных на выявление носителей. Важно помнить, однако, что эта информация сама по себе недостаточна для генетического консультирования, поскольку окончательный риск существенно зависит от конкретной родословной. Необходимы оба этапа этой процедуры, что нередко упускается из виду теми, кто для генетического консультирования использует только лабораторную информацию. Одно и то же отклонение от нормы, выявленное при лабораторной диагностике, можно трактовать по-разному в зависимости от вероятности носительства, установленной по родословной.
На практике сначала определяют вероятность носительства на основе информации о родословной. Полученное значение выражают в форме шансов или отношения правдоподобий (шансы = р: (1 - р), где p - вероятность. Пример: если р = 1/4, то шансы будут 0,25:0,75 = 1:3). На основе эмпирической информации о значениях креатинкиназы у носителей и в норме (таких, как в табл. П.8.2) устанавливаются шансы носительства при данном значении лабораторного теста. Шансы, полученные на основе обследования родословной и на основе лабораторной информации, перемножаются и превращаются в действительную вероятность или риск.
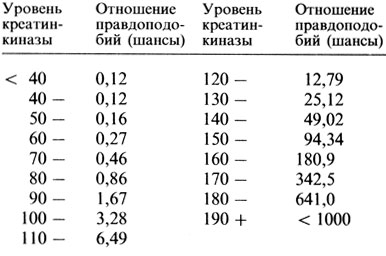
Таблица П.8.2. Вероятность носительства по гену мышечной дистрофии Дюшенна при разных уровнях сывороточной креатинкиназы*
* (Приведенные в таблице данные основываются на верхнем пределе (95% от нормы 100 ед./л среди взрослых женщин). Отношение правдоподобий представляет вероятность того, что данный показатель креатинкиназы для взрослой небеременной женщины получен из популяции точно установленных носителей против контрольной популяции (100 ед./л), где шансы носительства 3,28:1. Отметим, что при составлении этой таблицы тестировались равные по численности группы нормальных женщин и очевидных носителей. Действительный риск зависит от генетического риска, рассчитываемого по родословной (см. текст).)
Пример: сестра больного мужского пола с мышечной дистрофией Дюшенна, у которого поражен дядя (рис. П.8.2). Среднее значение креатинкиназы сестры на основе трех измерений составляет 100 ед./л (95% от нормы для взрослых женщин).

1 (Шансы основываются на реальных лабораторных данных.)
Вероятность носительства = а/(а + b) = 3,28/(3,28 + 1) = 3,28/4,28 = 0,77 = 77%,
где а - шансы носительства,
b - шансы неносительства.
Шансы, что эта женщина является носителем, увеличились с 50 до 77% за счет данных по креатинкиназе. Отметим, что вероятность женщине быть носителем при этом же самом значении фермента (= 100) составила бы лишь 14%, если ее генетический риск (вычисленный на основе информации из родословной) был 1:20. Этот риск все же намного выше, чем если бы семейных данных не было вовсе. В этом случае риск носительства равен 1/2000 (частота носителей в общей популяции), а действительный риск (основанный на лабораторном значении 100) составил бы 0,0016 или 1/628.
Как отличить здоровых людей от носителей с помощью лабораторных тестов. Поскольку с помощью лабораторного показателя мы не можем это сделать однозначно (т. е. не существует такого значения, выше или ниже которого мы могли бы с уверенностью говорить о норме или носительстве), следует учитывать, что, например, в случае мышечной дистрофии Дюшенна около одной трети носителей имеют "нормальный" уровень креатинкиназы (т. е. ниже двух стандартных отклонений от популяционной средней). Так, если из данных по родословной риск быть носителем составляет 1/2, а уровень креатинкиназы "нормальный", то суммарный риск можно оценить как 1/4, т. е. 25% (рис. П.8.6):

Наоборот, окончательный риск носительства можно вычислить как апостериорную вероятность: 1/6/(1/6 + 3/6) = 1/4 (см. также рис. П.8.6).
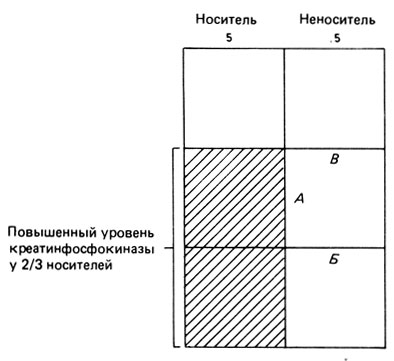
Рис. П.8.6. Уровень креатинкиназы при мышечной дистрофии Дюшенна. Пример: информация из родословной дает риск носительства 0,5 и неносительства 0,5. Линия (А) делит общее 'пространство' вероятностей на две равные по размеру части. Поскольку две трети носителей имеют повышенный уровень креатинкиназы, то вероятностное пространство делится линиями (Б) и (В) на три равные части. Носители, которые имеют аномальный уровень фермента, локализуются в области с косой штриховкой (это 2/3 носителей). Среди тех, кто имеет нормальные уровни креатинкиназы, один из четырех (белые квадраты) будет носителем
Действительный риск может оказаться ниже, если значения креатинкиназы располагаются в более низкой области нормального диапазона. Риск: может быть оценен выше, если соответствующие показатели относятся к области более высоких значений. На определенном уровне креатинкиназы (где шансы носитель/неноситель равны 1:1) этот показатель не будет влиять на риск быть носителем.
Использование информации о ПДРФ. Окончательную оценку риска можно еще улучшить, если использовать феномен полиморфизма по длине рестрикционных фрагментов (ПДРФ). Три соответствующих сайта расположены со стороны 5-'и 3'-концов гена мышечной дистрофии Дюшенна. Известно, что 45% британских женщин гетерозиготны по крайней мере по одному из них. Поскольку каждый из этих ДНК-маркеров расположен далеко от гена дистрофии (13-20 см), то кроссоверы между маркерным геном и геном мышечной дистрофии Дюшенна будут относительно частыми. Добавление данных по ПДРФ к информации, получаемой из родословной и при определении уровня креатинкиназы, часто снижает риск.
Пример использования всей информации приведен на рис. П.8.7. У женщины III, 2 имеется брат и умерший дядя, страдавший при жизни мышечной дистрофией Дюшенна. Она хочет знать, является ли она носителем. Показатель креатинкиназы 75 ед./л, тогда как у ее матери и бабушки со стороны матери эти значения равны НО и 125 ед./л соответственно. Ее риск быть носителем, рассчитанный по родословной, составляет 50:50. Исходя из уровня креатинкиназы, ее шансы быть носителем ранвы 0,46:1 (табл. П.8.2). Сначала вычисляется вероятность носительства по родословной и исходя из показателя креатинкиназы

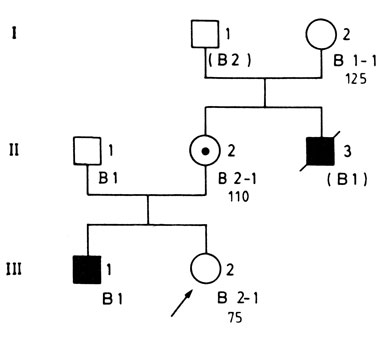
Рис. П.8.7. Родословная, информативная в отношении ПДРФ при мышечной дистрофии Дюшенна. В1 относится к более частому аллелю, а В2 - к более редкому аллелю этого локуса. (В2) и (В1) указывают на генотип. Числа под символами относятся к уровням креатинкиназы. Больной III, 1 получил аллель В1 от матери-носительницы (II,2), которая получила его от своей матери (I,2). III,2, которая имеет 50%-ный риск носительства, получила нормальный аллель В2 от матери и нормальный аллель В1 от отца. Следовательно, III,2 не унаследовала аллель мышечной дистрофии, если только в результате кроссинговера мутантный ген не попал в хромосому, несущую аллель В2 (вероятность 15%)
Данные о ПДРФ ее семьи (рис. П.8.7). свидетельствуют о том, что эта женщина (III, 2) унаследовала нормальную Х-хромосому (В2) от матери, хотя наблюдаемая картина могла быть следствием кроссинговера. С учетом расстояния между маркером и соответствующим геном ее шанс быть носителем (если основываться лишь на информации о ПДРФ) составляет 15%, т. е. шанс кроссовера. Объединение всей информации снижает риск носительства для нее до 1,4%, а риск иметь пораженного сына до 0,7%.
Появляется все больше данных о ПДРФ для гемофилии А и В. Оказалось, что практически все женщины гетерозиготны по пробе (специфической к гену или сегменту Х-хромосомы) для гемофилии А. Если по структуре родословной мы не можем сделать какого-либо вывода, вполне корректна постановка диагноза носительства гемофилии А с помощью ПДРФ (рис. П.8.8, П.8.9).
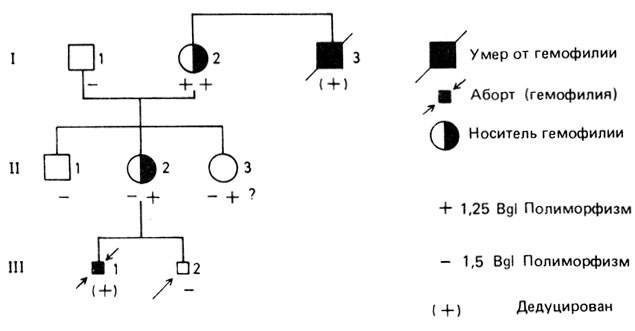
Рис. П.8.8. А. Принцип идентификации гетерозигот и пренатальная диагностика гемофилии А. Мать (I,1) является двойной гетерозиготой по аллелю гемофилии и ПДРФ-маркеру +. Б. Отец (I,1) здоров и имеет маркер -. Поскольку сын (II,2) унаследовал аллель гемофилии и маркер + от матери, то маркер + должен располагаться на той же хромосоме, что и аллель гемофилии (= фаза притяжения, разд. 3.4). Поскольку дочь (II,1) гетерозиготна +/-, она унаследовала хромосому, содержащую маркер + и ген гемофилии, от матери; она должна быть гетерозиготой по гемофилии. Плод (П,3) имеет маркер +, он должен унаследовать ту же хромосому и быть гемофиликом
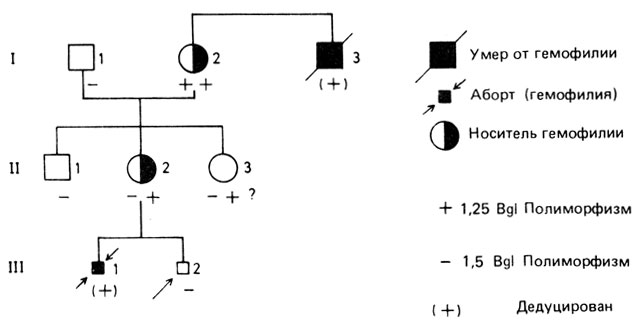
Рис. П.8.9. Родословная с гемофилией A (Din et al., 1985). Отметим, что гемофилию А можно было выявить у плода (III,2) посредством пренатальной диагностики, используя пробу фактора VIII и рестриктазу Bgl, без тестирования ДНК-фенотипов двух гемофиликов (III, 1 и I,3). ДНК-вариант у плода (III,2) идентичен таковому у здорового деда по материнской линии (I,1), что свидетельствует о том, что плод нормален. Отметим, что на основании анализа ДНК-варианта нельзя установить носительство сестры (II,3)
Повторный риск для детей непораженных носителей гена при аутосомно-доминантном наследовании. Иногда клинически непораженный индивид, у которого есть родственники с аутосомно-доминантным заболеванием со сниженной пенетрантностью, хочет получить совет относительно риска заболевания для своих детей. Независимо от точного значения пенетрантности было показано, что риск для детей индивида с 50%-ным риском никогда не превысит 9% [2366]. Причина этого заключается в том, что непораженный родитель вряд ли является носителем гена заболевания с высокой пенетрантностью. Наоборот, для болезней с низкой пенетрантностью, даже если родитель является носителем гена, вероятность для ребенка быть клинически пораженным мала.
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'