3.1.2. Аутосомно-доминантный тип наследования
Первое описание родословной с аутосомно-доминантным наследованием аномалии у человека дано в 1905 г. Фараби [656]. В учебниках это заболевание обычно называют брахидактилией (короткопалость), но из оригинальной статьи видно, что у больных не только укорочены фаланги пальцев рук и ног, но и редуцировано число самих фаланг (рис. 3.2). Такие люди характеризуются, кроме того, низким ростом (в среднем 159 см у трех мужчин), по-видимому, вследствие укорочения ног, а также короткими руками. Во всем остальном, по мнению Фараби,
"они совершенно нормальны и, по-видимому, почти не испытывают неудобств от своего порока. На свой единственный недостаток - короткие пальцы - жаловались лишь женщины-пианистки: они были не в состоянии охватить полную октаву и поэтому не могли хорошо играть".
![Рис. 3.2. Брахидактилия у одной из представительниц младшего поколения родословной Фараби. [708]](pic/000126.jpg)
Рис. 3.2. Брахидактилия у одной из представительниц младшего поколения родословной Фараби. [708]
На рис. 3.3 показана родословная с 36 пораженными (в поколениях II-V), среди которых 13 мужчин и 23 женщины. Среди непораженных - 18 мужчин и 15 женщин. Признак передается от одного из родителей примерно половине детей, причем передача признака не зависит от пола. К сожалению, Фараби не включил в родословную детей непораженных родственников. Анализ других родословных свидетельствует об отсутствии брахидактилии среди потомства родителей, которые не являются носителями доминантного гена. Впоследствии семья, о которой сообщал Фараби, была обследована вновь [708]. Добавились дети непораженных членов семьи и некоторых пораженных. Рентгенологическое обследование подтвердило, что укорочены не только кисти и стопы, но также и кости дистальных отделов верхних и нижних конечностей. Основной дефект затрагивает предположительно эпифизарные зоны роста. В настоящее время такая аномалия называется брахидактилией типа А1 (11250). (Число в скобках указывает порядковый номер заболевания по каталогу Мак-Кьюсика [133].)
![Рис. 3.3 Родословная с брахидактилией [656]. Обозначения: черные фигуры - пораженные женщины (●) и мужчины (■); римские цифры - номера поколений; арабские цифры: под фигурами - последовательные номера в родословной, внутри фигур - количество детей](pic/000127.jpg)
Рис. 3.3 Родословная с брахидактилией [656]. Обозначения: черные фигуры - пораженные женщины (●) и мужчины (■); римские цифры - номера поколений; арабские цифры: под фигурами - последовательные номера в родословной, внутри фигур - количество детей
Пораженные являются гетерозиготами по аутосомному гену, который имеет четкое и регулярное фенотипическое выражение. Следовательно, брахидактилия - признак доминантный. У данной семьи выявлены две особенности, которые позже оказались широко распространенными.
- Описанные аномалии были почти идентичными у всех членов семьи, и у каждого пораженного они обнаруживались на всех четырех конечностях. Это обычное свойство пороков с регулярным типом наследования. Причина симметрии очевидна, если предположить, что одни и те же гены действуют на все четыре конечности.
- Аномалия почти не сказывалась на здоровье пораженных. Отсутствие существенного ухудшения здоровья типично для случаев с обширными родословными. Репродуктивная функция у пораженных брахидактилией не нарушается, иначе признак не наследовался бы и вскоре после появления первичной мутации исчез. Это и есть ответ на вопрос, почему, особенно в случае более серьезных доминантных поражений, обширные родословные - скорее исключение, чем правило. Большинство заболеваний, вызываемых генными мутациями и наблюдаемых в нынешних поколениях, имеют сравнительно недавнее происхождение; часто они являются результатом вновь возникших мутаций в гаметах одного из родителей (разд. 5.1.3).
Позднее проявление (манифестация), неполная пенетрантность и варьирующая экспрессивность. Иногда тяжелое доминантное заболевание проявляется только во время или после репродуктивного периода. В этом случае, несмотря на тяжесть заболевания, обычно можно составить обширные родословные. Классический пример-это хорея Гентингтона (14310) -дегенеративное заболевание нервных клеток в базальных ганглиях, приводящее к непроизвольным движениям экстрапирамидного характера, изменениям личности и постепенно нарастающему слабоумию.
Вендт и Дром [941] провели всестороннее исследование всех случаев хореи Гентингтона в Западной Германии. Распределение этих случаев по возрасту начала заболевания показано на рис. 3.4. Подавляющая часть пораженных вступила в брак, когда у них уже проявились клинические симптомы. Даже среди нескольких тысяч пораженных авторы не смогли найти ни одного случая, который достоверно можно было бы отнести за счет новой мутации. Сходные результаты были получены в другом исследовании, проведенном в штате Мичиган (США) [850].
![Рис. 3.4. Распределение 802 случаев хореи Гентингтона по возрасту начала заболевания [941]](pic/000129.jpg)
Рис. 3.4. Распределение 802 случаев хореи Гентингтона по возрасту начала заболевания [941]
Другой характерный для доминантных признаков феномен-это неполная пенетрантность [912]. Пенетрантность - это статистическое понятие, которое определяется как доля индивидов с конкретным генотипом, у которых соответствующий этому генотипу фенотип проявляется. В случае неполной пенетрантности при передаче признака иногда одно поколение пропускается, причем признак не проявляется у того индивида, который, судя по родословной, должен быть гетерозиготным. При этом доля пораженных среди сибсов (после соответствующих поправок, разд. 3.3) оказывается меньше ожидаемой сегрегационной частоты. Примером заболевания с неполной пенетрантностью может служить ретинобластома (18020) - злокачественная опухоль глаз у детей. Двусторонние случаи (и случаи по крайней мере с двумя первичными опухолями) всегда наследуются доминантно, между тем большинство односторонних единичных опухолей не имеет наследственной природы и, вероятно, вызывается соматической мутацией (разд. 5.1.6). Даже в тех родословных, которые демонстрируют регулярное доминантное наследование, нередко можно обнаружить пропуск поколения (рис. 3.5). Так, при оценке сегрегационной частоты в одной большой выборке оказалось, что поражены около 45% сибсов вместо 50%, ожидаемых при регулярном доминантном наследовании. Следовательно, пенетрантность всех случаев (односторонних и двусторонних) составляет около 90%. Пенетрантность в семьях с двусторонними случаями выше, чем с односторонними. Следует, однако, учитывать, что оценка пенетрантности часто зависит от применяемых методов обследования.
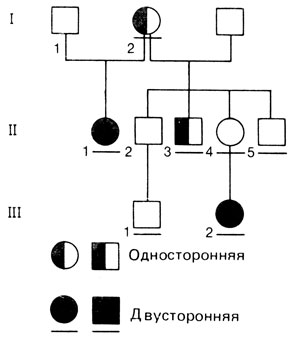
Рис. 3.5. Родословная с ретинобластомой в случае неполной пенетрантности. Непораженная женщина II. 4 должна быть гетерозиготой, поскольку ее мать I, 2 и дочь III. 2 поражены. (Черточка под фигурой означает, что больной обследован авторами.)
Для многих доминантных признаков характерна такая ситуация, что ген проявляется у всех гетерозигот, но в разной степени. Один из примеров - нейрофиброматоз (16220). У некоторых больных имеются отчетливо выраженные фиброматозные опухоли и "кофейные" пятна, а у других, даже в тех же семьях, можно обнаружить лишь пятна. Для описания этого явления используется термин "варьирующая экспрессивность" (Тимофеев-Ресовский, 1931, [912]) или равнозначный ему - "степень выражения" (manifestation rate). Однако следует помнить, что они не объясняют биологический механизм, а скорее служат указанием на то, чего мы не знаем, но что не следует игнорировать.
В самом деле, на первый взгляд кажется неожиданным, что так много доминантных заболеваний обнаруживают значительную межиндивидуальную изменчивость и по возрасту начала, и по тяжести проявления. Было бы понятно, если бы такие различия имели место только для разных семей. Молекулярно-биологические данные (разд. 5.1.4) говорят о том, что мутации, вызывающие эти заболевания, почти всегда отмечаются в разных семьях. Внутри семьи, как правило, наблюдается корреляция по возрасту начала и тяжести проявления заболевания. Например, для возраста начала хореи Гентингтона Вендт и Дром [941] получили коэффициент корреляции 0,57. Однако и внутри семей, в которых аномальные гены идентичны по происхождению, регистрируется заметная изменчивость. Когда в этих случаях мы апеллируем к таким понятиям, как "генетический фон" (или действие всех других генов), это снова не более чем указание на наше незнание. Анализ, основанный на методах формальной генетики, внес очень скромный вклад в понимание этих феноменов (обсуждение аллельной модификации и ограниченных полом генов-модификаторов см. в разд. 3.1.7).
Влияние гомозиготности на выражение аномальных доминантных генов. Аномальный ген считается доминантным, если фенотип гетерозигот четко отличается от фенотипа здоровых гомозигот. В популяциях человека почти все носители доминантных заболеваний гетерозиготны по той или иной мутации. Иногда случается, что два носителя одной и той же аномалии вступают в брак и имеют детей. Тогда четверть из них будут гомозиготами по мутантному аллелю. Такая ситуация вполне реальна, когда супруги-родственники. В кровнородственном браке между двумя носителями брахидактилии средней степени тяжести (11260) родился ребенок, у которого недоставало пальцев на руках и ногах и, кроме того, имелись множественные уродства скелета. Он умер в возрасте одного года. Однако у его сестры (как и у родителей) наблюдалась аномалия пальцев только средней степени тяжести [792].
Еще одним примером может служить пельгеровская (Pelger-Huet) аномалия нейтрофилов (16940) [823]. Это безвредная аномалия полиморфноядерных гранулоцитов, при которой вместо нормальной множественной сегментации ядер обнаруживаются только два сегмента примерно равного размера. Хроматин выглядит грубым и сморщенным. Эта аномалия четко наследуется по регулярному аутосомно-доминантному типу. Она не очень редкая (частота около 1:1000 или 1:3000 в центрально-европейских популяциях). Такой же дефект нейтрофилов обнаружен и у кролика. В связи с этим появилась потенциальная возможность получать в соответствующих скрещиваниях гомозиготное потомство, но ее реализация столкнулась с трудностями: гомозиготы часто погибали еще в пренатальном периоде. В конце концов удалось получить несколько особей, сохранявших жизнеспособность относительно короткое время. У этих животных все ядра гранулоцитов оказались круглыми, сегментации вообще не было, а хроматин выглядел грубым даже в лимфоцитах и плазматических клетках (рис. 3.6). Помимо гематологических аномалий были, однако, и другие симптомы, такие, как хондродисплазия конечностей и ребер и общая задержка развития. Аномалии ребер приводили, по-видимому, к сжатию органов грудной клетки и гибели животных. Нахтсхейм предсказал ту же самую аномалию лейкоцитов и подобные костные симптомы у гомозиготных людей. К счастью, это предсказание оказалось справедливым лишь отчасти.
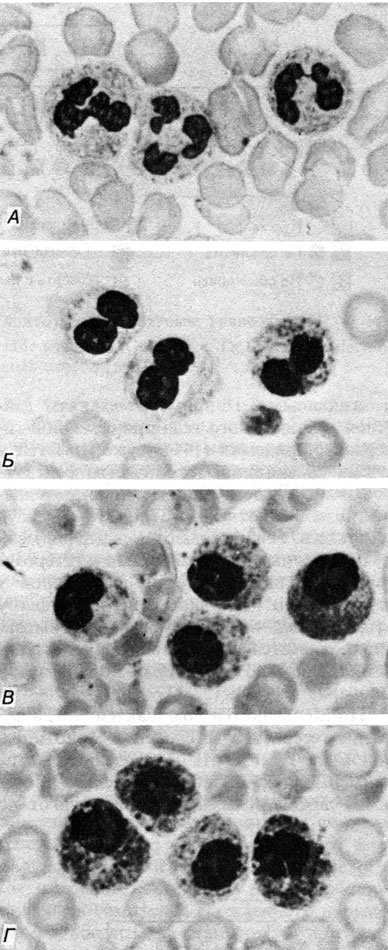
Рис. 3.6. Пельгеровская аномалия полиморфно-ядерных нейтрофилов. А. Нормальные гранулоциты. Б. Пельгеровские клетки у гетерозиготного носителя. В и Г. Гранулоциты у гомозигот человека и кролика соответственно
Первый случай гомозиготности у человека при синдроме пельгеровской аномалии лейкоцитов описан в 1952 г. в Голландии [707]. Это был ребенок от брака кузенов, цыган по происхождению. 94% его гранулоцитов оказались сходными с гранулоцитами гомозиготных кроликов (рис. 3.6, 3.7). Однако у этого гомозиготного индивида (как и у других) не было никаких признаков аномалии скелета.
![Рис. 3.7. Родословная с девочкой, гомозиготной по гену пельгеровской аномалии нейтрофилов [707]](pic/000132.jpg)
Рис. 3.7. Родословная с девочкой, гомозиготной по гену пельгеровской аномалии нейтрофилов [707]
Известны и другие примеры гомозиготности доминантных аномалий. В одной семье родители с наследственной гемморагической телеангиэктазией имели ребенка с множественными тяжелыми внутренними и внешними телеангиэктазами, умершего в возрасте 2,5 месяца [885]. Аналогично очень тяжелая форма буллезного эпидермолиза наблюдалась у двух из восьми детей, родители которых страдали той же болезнью, но в форме средней тяжести. Описаны также супруги, оба с миопатией, при которой поражаются дистальные мышечные группы конечностей. Они имели 16 детей, у троих из которых выявлены атипичные и особо тяжелые симптомы: при более ранней манифестации были поражены длинные сгибательные мышцы и проксимальные мышечные группы бедер [940]. Эпителиома типа adenoides cysticum (13270)-доминантное кожное заболевание, характеризующееся множественными узловатыми новообразованиями. Одна больная женщина, оба родителя которой страдали этим же заболеванием, имела особенно тяжелые симптомы, и все ее восемь детей проявили эту аномалию (рис. 3.8) [677]. Другими примерами могут служить ахондроплазия (10080) и синдром Элерса-Данлоса (13000) [832]. Все эти случаи указывают на то, что гомозиготы по доминантным аномалиям поражены тяжелее, чем гетерозиготы.
![Рис. 3.8. Гомозиготная женщина с кистозно-аденоидной эпителиомой и ее потомство в двух браках [677]](pic/000133.jpg)
Рис. 3.8. Гомозиготная женщина с кистозно-аденоидной эпителиомой и ее потомство в двух браках [677]
С точки зрения наших знаний о действии генов подобные факты не являются неожиданными. Например, механизм действия доминантного гена при семейной гиперхолестеринемии (14440) уже известен и связан со снижением количества рецепторов, взаимодействующих С липопротеинами низкой плотности. При этом, как и ожидалось, обнаруживаются различия между пораженными гомозиготами и гетерозиготами: полное отсутствие рецепторов у первых и 50%-ное снижение их числа у вторых (разд. 4.6.4). У пораженных гомозигот развивается массивная гиперхолестеринемия, и обычно они умирают от инфаркта миокарда до 30 лет.
По Менделю ген доминантен, когда фенотип гетерозиготы сходен с фенотипом одной из гомозигот. Приведенные выше примеры клинически более тяжелого проявления доминантных генов у гомозигот по сравнению с гетерозиготами показывают, что в генетике человека это строгое определение не выполняется. У человека доминантными называют все те признаки, по которым гетерозиготы заметно отличаются от нормальных гомозигот безотносительно к фенотипу аномальных гомозигот. В строго менделевском определении большинство или даже все доминантные признаки человека были бы "промежуточными". Однако в настоящее время общеупотребительно менее строгое толкование термина "доминирование".
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'