3.6.2. Мультифакториальное наследование в комбинации с пороговым эффектом
3.6.2.1. Описание модели: эксперименты на животных
В предшествующем разделе генетический анализ количественного признака на биометрическом уровне обсуждался в отношении нормальных признаков с унимодальным и почти нормальным распределением в популяции. Было показано, что простая модель аддитивною полигенного наследования удовлетворяет этим свойствам, и тем самым корреляции родитель-ребенок и сибс-сибс можно использовать для оценки наследуемости.
Однако для многих болезней и врожденных пороков развития наблюдаются четкие альтернативные распределения: индивид либо страдает данным заболеванием, либо нет. Между тем ни семейные исследования, ни изучение хромосом не смогли выявить какой-либо простой тип наследования или наличие хромосомной аномалии. Единственное, что следует из семейных данных, - это возрастание эмпирического риска для близких родственников оказаться пораженными тем же заболеванием (семейное накопление). Патофизиологические исследования позволяют предполагать сложный комплекс причин. В некоторых случаях очевидно влияние различных дополнительных биологических факторов. Осложнения привносят и средовые факторы: неправильное питание, инфекции и неизвестные агенты. Когда все эти генетические и средовые факторы вместе превышают определенный порог, способность организма сопротивляться оказывается ослабленной и индивид заболевает или умирает.
Термины "порог" и "подверженность" часто используются при обсуждении мультифакториального наследования. Порог подразумевает наличие резкого качественного различия: за этим порогом на шкале подверженности располагаются пораженные индивиды. Хотя понятие порога полезно для моделей мультифакториального наследования, вряд ли он на самом деле физически существует. Концепция подверженности подразумевает градуированный континуум возрастающей восприимчивости к заболеванию. Эта концепция сложнее аналитически, но с биологической точки зрения она, вероятно, применима к большинству ситуаций.
При редких заболеваниях с простым типом наследования мутация в единичном гене нарушает его функцию. В других случаях мутация приводит к трудностям лишь при особых обстоятельствах, как, например, при моногенно детерминированных реакциях на лекарства. Большинство признаков, однако, настолько сложны, что прямой анализ всех факторов оказывается практически невозможным, поскольку в подверженность вовлечено, вероятно, множество разных генов. Мы опять оказываемся в ситуации "черного ящика" - генетический анализ проще провести статистическими, нежели биологическими методами.
Генетические предсказания на таком сложном уровне должны основываться на нескольких предположениях:
- подверженность к заболеванию распределена более или менее нормально, и распределение имеет одну моду;
- подверженность обусловлена большим числом генов, действующих аддитивно, и каждый из них вносит равный вклад;
- когда подверженность превышает определенный порог, индивид заболевает или у него появляются нарушения. Хотя порог может быть четко определен, в большинстве случаев существует некоторая пороговая область, внутри которой от дополнительных средовых факторов будет зависеть, заболеет индивид или нет. Если описывать это в тех терминах, которые были введены выше, то можно сказать, что наследуемость меньше единицы (рис. 3.58).
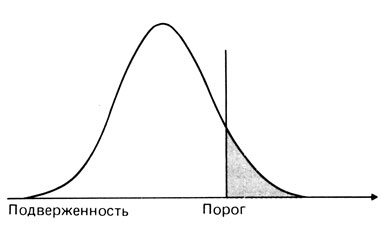
Рис. 3.58. Мультифакториальное наследование в сочетании с пороговым эффектом - простейшая ситуация. Подверженность заболеванию в общей популяции имеет нормальное распределение; индивиды справа от порога поражены болезнью
Очевидно, что эта модель слишком упрощает реальность, но она может быть полезна для понимания природы ряда широко распространенных заболеваний и пороков развития.
Эксперименты на животных. В экспериментальной генетике млекопитающих наследование некоторых признаков, например полидактилии у морской свинки (Райт [961]), объясняли пороговым эффектом. Скрещивались две линии: представители одной имели три пальца на задних конечностях (норма), у животных другой линии было четыре пальца (морфологический вариант). В поколении F1 обнаружено лишь несколько особей с четырьмя пальцами, тогда как во втором поколении (в потомстве скрещивания F1×F1) этот признак имелся примерно у четверти всех особей. Генетический анализ предположительно указывал на то, что скрещиваемые линии различаются по набору диаллельных систем с аддитивным эффектом четырех локусов: любое животное могло нести максимум восемь и минимум ноль положительных аллелей. При скрещивании двух гомозиготных линий (8×0) (рис. 3.59) гетерозиготное поколение F1 должно иметь четыре положительных аллеля. Такой генотип приводит к четырехпалым задним конечностям только в исключительных случаях. В поколении F2(F1×F1) присутствуют все комбинации положительных аллелей, что дает непрерывное распределение. В этом случае было принципиально показано, что аддитивное действие генов на самом деле может быть связано с пороговым проявлением (рис. 3.59). В другом примере удалось продемонстрировать не только дискретное, но и непрерывное фенотипическое выражение количественно варьирующей подверженности. Грюнеберг (1952) [690, 691] анализировал такую систему у мыши. В инбредной линии СВА у многих особей отсутствует третий молярный зуб: у 133 из 744, т. е. по крайней мере у одной мыши из пяти. Однако в черной линии С57 этот моляр почти всегда имеется. Скрещивание двух линий (СВА×С57) обнаружило, что тип наследования не является простым, несмотря на то что признак (зуб присутствует или отсутствует) обнаруживает четко выраженное альтернативное проявление. Даже у животных линии СВА с лишним зубом его размер был в среднем намного меньше, чем в черной линии С57 (рис 3.60). Следовательно, у животных линии СВА размер зуба варьирует непрерывно вплоть до определенного минимального порогового размера. Ниже этого порога зуб не формируется вовсе. Грюнеберг назвал это явление "квазинепрерывной изменчивостью". Сам по себе порог не очень четкий, правильнее говорить о некоторой пороговой области. Мультифакториальность генетической системы очевидна лишь при сопоставлении явных различий между двумя линиями и в скрещиваниях между ними. Внутри генетически однородной линии СВА изменчивость обусловлена только средовыми факторами.
![Рис. 3.59. Мультифакториальное наследование в сочетании с порогом - наличие лишнего пальца у морских свинок. Две родительские линии: одна с тремя пальцами, другая с четырьмя. Часть гибридов поколения F1 имеет четыре пальца. Генетический анализ выявил восемь аллелей, ответственных за этот признак. Частота особей, у которых имеется лишний палец, зависит от числа 'плюс' аллелей [961]](pic/000259.jpg)
Рис. 3.59. Мультифакториальное наследование в сочетании с порогом - наличие лишнего пальца у морских свинок. Две родительские линии: одна с тремя пальцами, другая с четырьмя. Часть гибридов поколения F1 имеет четыре пальца. Генетический анализ выявил восемь аллелей, ответственных за этот признак. Частота особей, у которых имеется лишний палец, зависит от числа 'плюс' аллелей [961]
![Рис. 3.60. Распределение размеров третьего нижнего моляра в двух инбредных линиях мышей: СВА (вверху) и С57 black (внизу) [690]](pic/000260.jpg)
Рис. 3.60. Распределение размеров третьего нижнего моляра в двух инбредных линиях мышей: СВА (вверху) и С57 black (внизу) [690]
Предпринимались неоднократные попытки продемонстрировать непрерывно распределенную подверженность и дискретные пороги у людей (см., например [619]), но в большинстве случаев имело место лишь дискретное проявление: "поражен" или "не поражен". Чтобы установить характер внутрисемейного распределения признака с пороговым проявлением в общем случае, рассмотрим теоретическую модель.
3.6.2.2. Простая теоретическая модель
Напомним модель, описанную в разд. 3.6.1: две пары аллелей с равными и аддитивными вкладами и частотами p1=p2=q1=q2=0,5. Предполагается, что эта генетическая система детерминирует подверженность. Заболевание проявляется, если в генотипе индивида имеются три или четыре плюс-аллеля (A или B) (рис. 3.61). Относительное число пораженных и непораженных детей в браках плюс × плюс (пораженный × пораженный), плюс × минус (пораженный × непораженный) и минус × минус (непораженный × непораженный) показано на рис. 3.62.
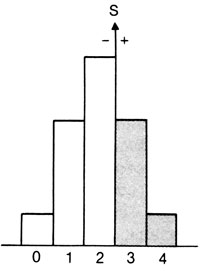
Рис. 3.61. Мультифакториальное наследование двух пар аллелей A, a и B, b в сочетании с порогом: распределение фенотипов (□ соответствует минус-фенотипу, ■ - плюс-фенотипу) в случайно скрещивающейся популяции. Частоты аллелей A=B=a=b=0,5. Возможны пять фенотипов (0, 1, 2, 3, 4)
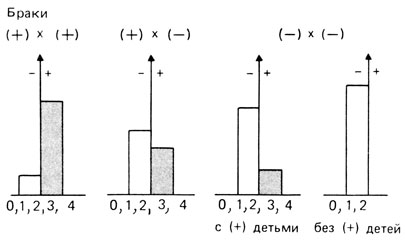
Рис. 3.62. Относительная частота детей (+) и (-) в четырех разных типах браков в соответствии с генетической моделью, описанной на рис. 3.61
Эти значения сильно напоминают частоты при простом аутосомно-доминантном типе наследования: для брака плюс × плюс они почти идентичны, если среди плюс-родителей предполагается определенное количество гомозигот. Для брака плюс × минус ожидаемые частоты почти совпадают, но для аддитивной модели они немного ниже. Все же регулярное доминирование с полной пенетрантностью у гетерозигот почти всегда четко отличимо от мультифакториального наследования, особенно если имеются данные по крайней мере о трех поколениях в семье. Однако при неполной пенетрантности проблема дискриминации от мультифакториального наследования с пороговым проявлением становится крайне затруднительной: в этом случае можно ожидать, что в некоторых сибствах оба родителя будут непораженными, а сегрегационное отношение окажется меньше 0,5. Тогда может помочь сравнение сибсов от браков плюс × минус и минус × минус. В мультифакториальной модели ожидается меньшая доля пораженных среди детей двух непораженных родителей по сравнению с сибсами, у которых один родитель поражен. При простом аутосомном доминировании и неполной пенетрантности сегрегационные отношения в обоих типах семей должны быть идентичны. Правда, этот аргумент можно оспорить, утверждая, что на пенетрантность повлиял генетический фон, но тогда проблема становится в значительной степени семантической: с самого начала было очевидно, что предположение о равном влиянии на проявление данного признака всех генов является сильным упрощением. Однако если вклад генов считать неравным, то начиная с какого уровня влияния одного локуса на фенотипическую изменчивость мы можем говорить об эффектах "главного гена"?
В приложении 4 будет рассмотрена более общая модель мультифакториального наследования признака с пороговым проявлением. Описываемые ниже критерии мультифакториального наследования, позволяющие отличить этот тип от простого диаллельного моногенного наследования, интуитивно следуют из описанной выше простой специальной модели, но их можно получить вполне строго из более общей модели, описанной в приложении 4.
3.6.2.3. Как нужно использовать модель для анализа данных [925]?
В анализе реальных данных теоретические результаты этого раздела следует использовать критически. Как уже неоднократно упоминалось, мультифакториальная модель является абстрактной и слишком упрощает сложную мозаику взаимодействия множества генов, формирующего подверженность. Кроме того, на практике обычно имеют дело с ограниченным объемом данных, что приводит к большой выборочной дисперсии.
Качественные (или полуколичественные) критерии мультифакториального наследования. Можно сформулировать четыре таких критерия.
1. Близнецовый критерий: если конкордантность монозиготных (МЗ) близнецов вчетверо выше, чем конкордантность дизиготных (ДЗ) близнецов, то мультифакториальная модель более адекватна, чем простая диаллельная модель (табл. 3.19). Обратное неверно: если отношение конкордантностей меньше четырех, то мультифакториальная гипотеза необязательно должна быть отвергнута.

Таблица 3.19. Конкордатность близнецов при разных типах наследования
2. Сегрегационное отношение пораженных и непораженных сибсов в браках плюс × минус и минус × минус: если доля пораженных сибсов в браках с одним пораженным родителем выше в 2,5 раза (и более), чем та же доля среди детей в браках с двумя непораженными родителями, то следует предпочесть мультифакториальную модель. Но и в данном случае надо помнить, что если указанное отношение меньше 2,5, то это еще не исключает мультифакториальное наследование.
3. Соотношение полов пораженных: многие признаки, в отношении которых следует рассматривать мультифакториальное наследование, обнаруживают половые различия по распространенности в популяции. В большинстве случаев лишь малая доля таких различий может быть обусловлена генами собственно половых хромосом. Основные источники этих различий связаны с физиологией пола. Если это так, то разумно предположить, что генотипическая компонента подверженности имеет одинаковое распределение для обоих полов, но пороги проявления признака различаются. Отсюда следует, что пораженные того пола, у которого более высокий порог проявления, будут иметь в среднем и более высокое индивидуальное значение по сравнению с индивидами другого пола. Система подверженности такого рода должна отражать различия в частоте пораженных родственников: у пробандов реже поражаемого пола больные родственники должны встречаться чаще, чем у пробандов чаще поражаемого пола (если, конечно, сравниваются одинаковые степени родства). Этот результат впервые был сформулирован Картером [601] и иногда называется эффектом Картера. Он был продемонстрирован на примере пилоростеноза - врожденной аномалии, при которой утолщение мышечного слоя пилоруса препятствует выходу содержимого желудка в двенадцатиперстную кишку. Хотя этот дефект встречается у новорожденных мальчиков чаще, чем у девочек, однако было показано, что среди родственников пораженных девочек пилоростеноз встречается чаще, чем среди родственников пораженных мальчиков (см. табл. 3.20 и рис. 3.65). Эффект Картера был продемонстрирован также для одной из особенностей электроэнцефалограммы (так называемые "диффузные" β-волны), которая встречается чаще у женщин, чем у мужчин. В этом случае родственники пробандов мужского пола намного чаще обнаруживают ту же особенность ЭЭГ, чем родственники пробандов женского пола [921]. С другой стороны, эффект Картера не удалось выявить для аномалий типа "заячьей губы" и "волчьей пасти", для которых, вообще говоря, ожидалось, что среди родственников пробандов-женщин будет больше пораженных, так как женский пол поражается в этом случае реже.
![Таблица 3.20. Пилоростеноз: частота среди близких родственников пробандов мужского и женского пола (Fuhrmann, Vogel, 1983 [71])](pic/000264.jpg)
Таблица 3.20. Пилоростеноз: частота среди близких родственников пробандов мужского и женского пола (Fuhrmann, Vogel, 1983 [71])
![Рис. 3.65. Мультифакториальное заболевание может быть более частым среди лиц определенного пола. Например, пилоростеноз чаще встречается у мужчин, чем у женщин. Можно предположить, что генетическая подверженность идентична для обоих полов, но положение порога на шкале подверженности разное. В результате пораженные мужчины в среднем проявляют признак с меньшей генетической подверженностью, чем в среднем пораженные женщины. Следовательно, частота такого признака среди родственников пробандов мужского пола ожидается ниже, чем среди родственников пробандов женского пола, которые несут больше генов предрасположенности, чем пораженные мужчины. Этот феномен иногда называют 'эффектом Картера' [601]](pic/000267.jpg)
Рис. 3.65. Мультифакториальное заболевание может быть более частым среди лиц определенного пола. Например, пилоростеноз чаще встречается у мужчин, чем у женщин. Можно предположить, что генетическая подверженность идентична для обоих полов, но положение порога на шкале подверженности разное. В результате пораженные мужчины в среднем проявляют признак с меньшей генетической подверженностью, чем в среднем пораженные женщины. Следовательно, частота такого признака среди родственников пробандов мужского пола ожидается ниже, чем среди родственников пробандов женского пола, которые несут больше генов предрасположенности, чем пораженные мужчины. Этот феномен иногда называют 'эффектом Картера' [601]
4. Кровное родство: в обсуждаемых моделях предполагается случайное скрещивание. Однако в случае кровного родства распределение подверженности в популяции характеризуется более высокой дисперсией:
Здесь F - коэффициент инбридинга, VF - дисперсия среди всего потомства в браках с коэффициентом инбридинга F, VO -дисперсия при случайном скрещивании (рис. 3.63). На рис. 3.64 показано увеличение частоты пораженных среди детей от браков двоюродных сибсов (F=1/16) относительно частоты пораженных в панмиксной популяции. Для сравнения представлено намного более выраженное увеличение частоты, наблюдаемое для моногенного аутосомно-рецессивного наследования. Однако в большинстве случаев более подходящей альтернативой мультифакториальной модели будет скорее аутосомно-доминантное наследование с неполной пенетрантностью, чем аутосомно-рецессивное. Следовательно, умеренное увеличение частоты признака, связанное с повышением уровня инбридинга, является дополнительным аргументом в пользу мультифакториальной модели против аутосомно-доминантной при условии, конечно, что примесь семей с редким аутосомно-рецессивным сходным признаком исключена.
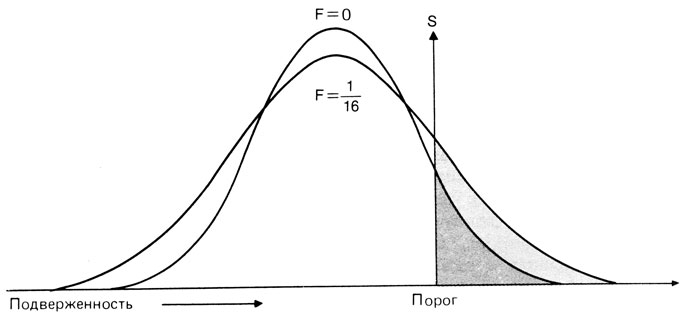
Рис. 3.63. Распределение генетической подверженности при случайном скрещивании и при F=1/16 (брак двоюродных сибсов). Области справа от порога указывают возрастание частоты порогового признака. S-порог
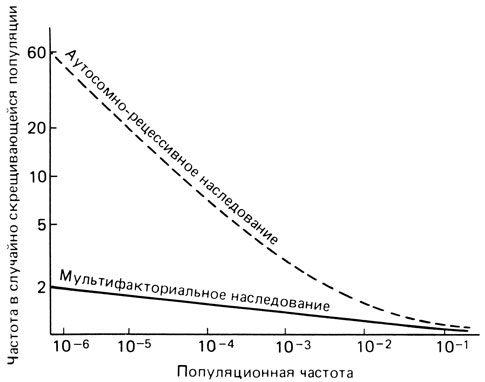
Рис. 3.64. Возрастание частоты аутосомно-рецессивных и мультифакториальных признаков среди детей в браках двоюродных сибсов в сравнении с популяционной частотой
Количественные критерии. Полуколичественные критерии, на которых основывается генетическая модель, не могут полностью удовлетворить нас. Необходим метод количественного сравнения. В этом случае на основе наблюдаемых частот в выборке можно вычислить 95%-ные доверительные интервалы для теоретически ожидаемых частот Q11, Q21 и Q22 среди детей браков минус × минус, плюс × минус и плюс × плюс и узнать, попадают ли ожидаемые значения в эти доверительные интервалы (приложение 4). Однако лучше, по-видимому, проверить, соответствует ли общее распределение всех частот среди разных типов родственников тем ожидаемым значениям, которые получаются на основе как одной, так и другой модели. Такие методы были описаны Мортоном и соавт. [804] и Смитом [875а]. (Смотрите, например, исследование по врожденной глаукоме [628].)
Сначала определяют частоту изучаемого признака среди родственников пробанда. Затем совместную вероятность всех этих частот сравнивают с соответствующими теоретически ожидаемыми значениями, с одной стороны, для мультифакториальной модели, а с другой - для диаллельной моногенной модели. Если одна из моделей дает результаты, содержащиеся внутри доверительных интервалов, а другая обнаруживает значимые отклонения, то принимается первая модель. Вычислительные аспекты будут рассмотрены в приложении 4. Ряд авторов [646; 852; 963] предложили сходные методы идентификации эффектов "главных генов".
3.6.2.4. Какой вывод следует сделать, если статистический анализ не дает четкого ответа?
Выше уже указывалось, что совместимость полученных данных с генетической моделью еще не означает, что эта модель истинная. Совершенно разные модели могут одинаково хорошо соответствовать одним и тем же данным. Как показано выше (а также в приложении 4), имеется существенное перекрывание ожидаемых значений в разных моделях, в частности в описанных здесь для примера диаллельной моногенной и мультифакториальной. Общее правило состоит в том, что гипотезу можно отвергнуть, если она не соответствует наблюдениям, но она и не может быть принята до тех пор, пока не исключены все другие возможные гипотезы. Однако специалист по генетике человека, часто имеющий дело с просто наследующимися аномалиями, нередко забывает это правило, поскольку в обычных условиях менделевского наследования между наблюдением и генетической гипотезой имеется вполне прямая связь.
Как следует поступать, когда статистические данные не позволяют выбрать какую-либо из этих гипотез? Наиболее очевидный ответ - оставить проблему открытой. Необходимо более тщательное изучение таких случаев.
Гипотеза главного гена обладает многими преимуществами с точки зрения стратегии исследования. Аномалия, обнаруживающая простой тип наследования, должна иметь и четкую биохимическую причину: недостаток или нарушение нормального генного продукта структурного или регуляторного гена. Принятие гипотезы главного гена естественным образом ведет к биохимическому анализу этой причины. Такие исследования для аутосомно-доминантных признаков остаются еще в очень зачаточном состоянии (разд. 4.6). Для мультифакториальных признаков, обусловленных комбинациями различных малых физиологических отклонений (действующих, вероятно, вместе со средовыми факторами), гипотеза главного гена обычно ни к чему не приводит и поэтому вызывает у исследователей только разочарование. Примером может служить исследование нейрофизиологических, биохимических и иммунологических основ шизофрении (разд. 8.2.3.7).
Модель мультифакториального наследования более осторожная и консервативная: применяя ее как аналитический аппарат первичного описания данных, мы осознаем, что она ограничена анализом на уровне, наиболее удаленном от действия гена, - "черный ящик" все еще нужно открывать. Размышляя о выборе стратегии, мы не должны двигаться лишь в одном направлении увеличения мощности генетической гипотезы, а должны оставлять открытыми и другие возможности. Если, следуя какой-нибудь одной из них, мы на самом деле приходим к открытию действия главного гена, это переводит наш анализ на более глубокий генетический или биохимический уровень. Однако если попытка не увенчается успехом, то мы все же увидим, как малое отклонение какого-то физиологического параметра (которое присутствует лишь у части пробандов) может взаимодействовать с другими малыми отклонениями и тем самым формировать истинно мультифакториальную подверженность заболеванию. Кроме того, с помощью других исследовательских стратегий, предназначенных для более тонкого изучения на уровне, более близком к действию гена, можно попытаться рассмотреть по крайней мере какую-то одну компоненту мультифакториальной системы.
Следовательно, если с помощью четкого генетического или биохимического (или обоих) критериев нельзя определенно установить действие единичного гена, то принятие более общей мультифакториальной модели является мудрым решением. Однако мы должны помнить, что на самом деле во многих случаях нельзя исключить главный ген. Это обстоятельство очень важно учитывать, особенно если речь идет об оценке генетического риска, связанного с мутагенными факторами (разд. 5.2.1): однозначное принятие мультифакториальной модели может привести к недооценке генетической опасности. Чтобы избежать этой ошибки, следует помнить о результатах некоторых экспериментов в генетических исследованиях млекопитающих.
3.6.2.5. Индуцированные радиацией доминантные мутации у мыши: мутации главных генов, не выявленные у человека
Экспериментальная работа с млекопитающими, физиология развития которых ближе всего к человеку, показывает, как действие главного гена может быть скрыто за фенотипической изменчивостью организма. Такие главные гены идентифицируются с помощью подходящих экспериментальных скрещиваний или на основе феногенетического анализа индуцированных мутаций. Мы обсудим один пример, который важен также для оценки риска индуцированных мутаций у человека [865; 640].
Генетические дефекты, связанные с доминантными мутациями, можно выявить путем сравнения потомков первого поколения от опытных и контрольных животных. Однако для многих признаков трудно провести различие между вновь возникшими мутациями и внутрилинейной изменчивостью. Эта трудность была преодолена для некоторых скелетных аномалий у мыши. В мутационном эксперименте аномалии, наблюдавшиеся в поколении F1, можно разделить на те, которые проявляются крайне редко на протяжении всего эксперимента (класс 1), и на такие, которые встречаются много чаще (класс 2). Разумная рабочая гипотеза (при исследовании многих сотен особей) заключается в предположении, что большинство очень редких аномалий (класс 1) имеют мутационное происхождение, тогда как большинство частых аномалий (класс 2) обусловлены внутрилинейной изменчивостью. Согласно этой гипотезе, мутагенные факторы типа ионизирующей радиации должны повысить количество первично очень редких (класс 1) аномалий. Это подтверждено в достаточном количестве экспериментов с ионизирующей радиацией и химическими мутагенами.
Фенотипически большинство редких (класс 1) и более распространенных (класс 2) аномалий представляют собой множественные минорные варианты скелета. Некоторые из них, например аномалии позвоночника, вредны в разной степени. Относительно 31 аномального варианта скелета с помощью экспериментальных скрещиваний было подтверждено, что они вызываются доминантными мутациями. Выделим две характеристики этих доминантных мутаций.
- Некоторые или все аномалии мутационного происхождения имеют низкую пенетрантность.
- Морфологически можно выявить лишь небольшую долю этих мутаций, и те, которые можно выявить, не проявляются у большинства носителей гена. Экспериментальные скрещивания показали, что в потомстве особей F1 вероятность выщепления аномальных фенотипов оказывается гораздо ниже ожидаемой 0,5.
Сопоставляя эти результаты с данными по доминантным мутациям у человека, можно предположить, что трудности выявления таких мутаций у мыши не столь актуальны для человека, тщательное медицинское обследование позволяет идентифицировать большинство этих мутаций. При рассмотрении доминантных генов с неполной пенетрантностью очень важной проблемой остается установление низкой пенетрантности аутосомных мутаций. Кроме того, часто регистрируемые аномалии обнаруживают поразительную степень изменчивости между особями, несущими мутантные гены, идентичные по происхождению. С другой стороны, фенотипические спектры одной и той же аномалии у носителей разных мутаций сильно перекрываются. Фенотипические проявления некоторых мутаций были почти идентичными. Для сравнения этих результатов с генетическими данными у человека необходимо было бы выявить у него уродства скелета, сходные с таковыми у мыши. Попытки такого рода уже предпринимались, но из-за неразработанности вопросов генетики скелетных аномалий человека большого успеха достичь не удалось. Недавний "всплеск" новых исследований в этой области [774], возможно, приведет к пересмотру накопленных данных и некоторых обобщений. Обескураживает, однако, то обстоятельство, что до сих пор не удается найти у мыши скелетные мутации, идентичные мутациям у человека. Описанные эксперименты оставили нерешенными некоторые вопросы, в частности вопрос о возможности незначительных хромосомных перестроек. В порядке рабочей гипотезы можно выдвинуть следующее предположение: у человека существует большое количество доминантных мутаций, вызывающих широкий диапазон морфологической изменчивости и, вероятно, оказывающих влияние на здоровье. Однако современные методы феногенетического анализа весьма несовершенны и не дают возможность раскрыть генетическую основу этой изменчивости.
3.6.2.6. Идентификация элементарных клинико-генетических вариантов моногенного наследования с использованием дополнительных фенотипических критериев
Иногда в пределах большой гетерогенной группы больных можно выделить отдельные формы патологии с отчетливо менделевским наследованием. Это удается сделать на основе детального клинического изучения, лабораторных исследований и генетического анализа. Данные, полученные при этом, позволяют отделить генетические случаи от негенетических. Подобные результаты были получены для умственной отсталости [2157], глухоты [669] и слепоты [670]. С развитием и совершенствованием нозологии в области психоневрологии и с повышением уровня клинических исследований некоторые задержки умственного развития, которые ранее относили к общей группе клинически недифференцированных форм, теперь можно достаточно четко классифицировать. В качестве примера весьма распространенного признака можно упомянуть X-сцепленную форму умственной отсталости с маркерной "ломкой" X-хромосомой [2220]. Успешными в этом смысле были также исследования слепых и глухих детей, живущих при лечебных учреждениях. Оказалось, что около 50% всех случаев глухоты и слепоты имели генетическую природу. И практически все эти случаи были скорее менделевскими, чем мультифакториальными. Среди них было найдено много разных клинических форм с простым типом наследования.
Почти всегда в рамках мультифакториального заболевания можно идентифицировать редкие менделевские варианты. Так, X-сцепленная недостаточность фермента HGPRT составляет 1% всех случаев подагры. Некоторые случаи гипертонической болезни вызываются редкой наследственной феохромоцитомой. Язвенная болезнь желудка и двенадцатиперстной кишки выступает как часть симптомокомплекса при болезни Золлингера-Эллисона. Рак пищевода иногда возникает при генетически обусловленных кератомах одновременно на ладонях и подошвах (рис. 3.66).
![Рис. 3.66. Рак пищевода как дополнительный симптом (■) у больных особой аутосомно-доминантной формой кератоматоза ладоней и подошв [716]](pic/000268.jpg)
Рис. 3.66. Рак пищевода как дополнительный симптом (■) у больных особой аутосомно-доминантной формой кератоматоза ладоней и подошв [716]
Имеется ряд синдромов, при которых рак оказывается частью более сложной плейотропной картины (разд. 5.1.6). Иногда в семьях наблюдается доминантное наследование более или менее распространенных форм рака. В этом случае раннее начало и множественные поражения помогают отделить эти проявления главного гена от обычных типов рака. В родословной на рис. 3.66 возраст проявления рака приходился на 34, 37, 38, 43, 44, 45, 46, 52 и 63 года. Однако все эти случаи, кроме последнего, очень необычны для рака пищевода. В дерматологии наблюдается множество как изолированных, так и семейных случаев доброкачественных и злокачественных опухолей. Установлено правило [84], согласно которому единичные опухоли у одного больного имеют негенетическое происхождение, тогда как множественные опухоли имеют тенденцию наследоваться, причем часто обнаруживают аутосомно-доминантный тип наследования (см. также раздел 5.1.6).
3.6.2.7. Как анализировать мультифакториальный признак, если отдельные формы с простыми типами наследования выделить нельзя?
Сложный функциональный дефект вызывается комбинацией малых нарушений. Как упоминалось выше, аддитивно-полигенная модель, используемая для анализа мультифакториального наследования, является слишком упрощенной абстракцией. В действительности изменчивость не одномерна, и к определенному заболеванию может привести совместное действие ряда разных генетически детерминированных физиологических отклонений. Желательно идентифицировать хотя бы некоторые из таких факторов.
В двух выборках детей с косоглазием, обследованных одним автором [856], были получены данные о родителях и сибсах (табл. 3.21). Из 12 пар монозиготных близнецов 11 были конкордантными, тогда как из 27 пар дизиготных близнецов конкордантными были лишь 7. Эти данные говорят о мультифакториальном наследовании. Нельзя, конечно, исключать и неполное доминирование, но для этого потребовалось бы постулировать влияние генетического фона.
![Таблица 3.21. Частота косоглазия среди сибсов пробандов с косоглазием (Richter, 1966) (Vogel, Krüger, 1967 [925]) (+ - манифестная форма)](pic/000269.jpg)
Таблица 3.21. Частота косоглазия среди сибсов пробандов с косоглазием (Richter, 1966) (Vogel, Krüger, 1967 [925]) (+ - манифестная форма)
Известно, что косоглазие является конечным результатом ряда незначительных физиологических отклонений. Каждое из них в отдельности можно преодолеть и восстановить нормальное зрение. Но когда проявляется целая совокупность таких нарушений, регуляторная способность зрительной системы декомпенсируется и появляется косоглазие. Показано, что такие нарушения часто обнаруживаются у близких родственников пробанда. В родословной на рис. 3.67 трое больных детей с косоглазием; двое родителей обнаруживают изолированную гетерофорию (легкую моторную недостаточность). Один родитель имел изолированную аномалию рефракции глаза, другой - гетерофорию. Выводы этого исследования о том, что косоглазие является мультифакториальным признаком и некоторые физиологические отклонения, вовлеченные в общую систему подверженности, можно идентифицировать, позже были подтверждены и расширены на примере изучения другой популяции [709].
![Рис. 3.67. Косоглазие у трех членов семьи. У других родственников обнаружены другие минорные аномалии. Точечными и штриховыми линиями обозначены различные пограничные случаи. Сенсорные аномалии, наблюдавшиеся в такой родословной, включали, например, амблиопию или несовершенное бинокулярное зрение [856]](pic/000270.jpg)
Рис. 3.67. Косоглазие у трех членов семьи. У других родственников обнаружены другие минорные аномалии. Точечными и штриховыми линиями обозначены различные пограничные случаи. Сенсорные аномалии, наблюдавшиеся в такой родословной, включали, например, амблиопию или несовершенное бинокулярное зрение [856]
Успешной оказалась также попытка дифференцировать генетическую компоненту подверженности при врожденном вывихе бедра. В этом случае было показано, что в подверженность вовлечены, вероятно, как аддитивно-полигенные факторы, определяющие поражение поверхности вертлужной впадины, так и моногенный фактор, обусловливающий общую слабость сочленения [619].
Семейные исследования, ориентированные на детальное изучение фенотипических проявлений с целью поиска сходных или ассоциирующих микроаномалий, несомненно могут помочь в понимании относительной важности отдельных элементов, приводящих в комбинации к сложному функциональному дефекту. Это возможно, даже если нельзя идентифицировать действие единичного гена.
Мультифакториальная система охватывает все факторы подверженности, которые могут привести к группе сходных заболеваний: отдельные формы проявляются при этом специфической комбинацией ряда факторов. Группа "атопических заболеваний" включает атопический дерматит, бронхиальную астму и сенную лихорадку. На рис. 3.68 приведены данные об относительной частоте пробандов с одной, двумя или тремя атопиями в популяции Цюриха [894]. В основном эти данные, включая семейные, совместимы с мультифакториальным наследованием. Можно, однако, поставить и такой вопрос, является ли генетическая компонента подверженности при атопических заболеваниях одномерной и количественной, или существуют другие генетические компоненты, отражающие органоспецифичность проявления заболевания?
![Рис. 3.68. Относительные частоты пробандов с одной, двумя или даже тремя атопическими болезнями. As - астма, Ri - ринит, At - атопический дерматит. (Данные из Цюриха, Швейцария; Schnyder, 1960 [894].)](pic/000271.jpg)
Рис. 3.68. Относительные частоты пробандов с одной, двумя или даже тремя атопическими болезнями. As - астма, Ri - ринит, At - атопический дерматит. (Данные из Цюриха, Швейцария; Schnyder, 1960 [894].)
Если подверженность имеет одномерное распределение, то кожные атопии (дерматиты) и дыхательные атопии (астма и сенная лихорадка) должны встречаться примерно с одинаковой частотой среди родственников пробандов как с кожными, так и с дыхательными атопиями. С другой стороны, если существует влияние органоспецифических факторов, то среди родственников пробандов должно наблюдаться определенное накопление сходных атопий.
Рис. 3.69 иллюстрирует результаты такого сравнения: среди родственников первой степени пробандов-астматиков дыхательные атопии встречаются чаще, тогда как среди родственников пробандов с дерматитами превалируют атопические дерматиты. Таким образом, в пределах мультифакториальной генетической системы, определяющей генетическую подверженность к атопическим заболеваниям, существуют как факторы общего характера, т. е. усиливающие общую подверженность при атопиях, так и действующие совместно с ними другие факторы, влияющие на поражение конкретных органов.
![Рис. 3.69. Частота атопических дерматитов и респираторных атопий у родственников пробандов с астмой (A), с атопическим дерматитом (B). As - астма, Ri - ринит, At - атопический дерматит [924]](pic/000272.jpg)
Рис. 3.69. Частота атопических дерматитов и респираторных атопий у родственников пробандов с астмой (A), с атопическим дерматитом (B). As - астма, Ri - ринит, At - атопический дерматит [924]
Подобный клинико-генетический анализ приемлем в большей степени, чем простые попытки подобрать значения общей частоты атопий, согласующиеся с ожидаемыми значениями в рамках генетической модели, которая исходно является сильно упрощенной. Все же, несмотря на такое усовершенствование, генетический анализ остается по своему характеру биометрическим, т. е. далеким от непосредственного действия гена. Теперь на очереди задача "вскрытия черного ящика". Так, можно показать, что аллергический насморк (сенная лихорадка) обусловлен взаимодействием двух генов, один из которых регулирует базальную продукцию IgE (иммуноглобулинов класса E), а другой влияет на продукцию IgE в реакции на конкретный аллерген. Второй из них идентичен или близко сцеплен с аллелем HLA-A2 [775]. Весьма возможно также, что существует генетический контроль на других уровнях иммунного ответа [844]. Представляет интерес вопрос о вероятном селективном преимуществе генотипов, связанных с атопическими заболеваниями, которое проявляется в более примитивных условиях жизни, что будет обсуждаться в разд. 6.2.1.
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'