Практический пример сегрегационного анализа с использованием большой выборки: полная глухонемота
Здесь мы рассмотрим на конкретном примере некоторые методы, описанные в разд. 3.3.6 и приложении 3. Стевенсон и Чизмен (1955) [899] собрали все случаи полной глухонемоты в Северной Ирландии. На момент обследования были живы 613 глухих, которые родились с этим дефектом или очень рано потеряли слух. Кроме того, у авторов имелись дополнительные данные о 85 лишенных слуха людях, которых уже не было в живых.
Регистрация семей. Сведения о глухонемых хранятся в архивах благотворительных или медицинских учреждений. Поскольку исследователи наладили контакт со всеми врачами в Северной Ирландии, регистрацию можно считать достаточно полной. Хотя несколько пробандов были зарегистрированы косвенно через пораженных родственников, даже в этих случаях в архивах были найдены записи одного или другого типа. А это означает, что в соответствии с данным выше определением, все пораженные могут считаться пробандами. Поскольку индивиды регистрировались по крайней мере двумя способами, можно предположить полный или усеченный отбор.
Семейные данные собирали в процессе личных визитов одного из авторов или их сотрудников. Их дополняли, насколько это возможно, физическими обследованиями и другими объективными данными. Здесь оказывались полезными записи, производимые в специальных школах относительно пораженных родственников, родство родителей и другие подобного рода данные. Частота признака составила 45 случаев на 100000 жителей.
Клинические аспекты. Клиническое обследование больных проводили для того, чтобы расширить знание о патогенезе и симптомах и исключить те средовые агенты, которые могли вызвать глухонемоту в раннем детстве (например, краснуха, эритробластоз, ототоксичные лекарства, перинатальная травма, энцефалит, менингит и отит). Однако часто на основе только клинических и аудиометрических данных невозможно было поставить диагноз. Результаты обследования в сочетании с историей болезни позволили исключить из выборки 183 живущих и 2 умерших больных.
Генетический анализ. Отдельно анализировали следующие три типа семей:
1) родители Г × Г,
2) родители Г × Н,
3) родители Н × Н
(Г - наследственная глухонемота, Н - непораженный).
Данные для третьей группы (Н × Н) приведены в табл. П.3.1. Предварительное исследование и большое число непораженных сибсов предполагают аутосомно-рецессивный тип наследования. Следовательно, используется описанный выше метод тестирования и наблюдаемые частоты сравнивают с их ожидаемыми значениями при полном отборе. Однако полученный результат не совместим с генетической гипотезой. Имеется высоко значимый недостаток пораженных (χ2 = 26,60 с 1 ст. св.).
Этот результат показывает несовершенство метода тестирования по сравнению с методом оценки. Если в первом случае мы просто получаем отрицательный ответ, то во втором мы оказываемся в состоянии оценить сегрегационное отношение в этих семьях.
Таким образом, целесообразно использовать метод оценки с исходными значениями р = 0,20 и q = 0,80 (уравнение П.3.1)
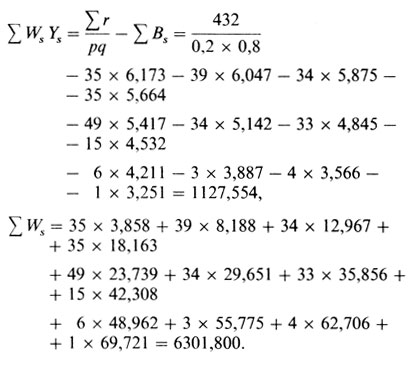
Это дает
| pˆ = | 1127,554 | = 0,17893 |
| 6301,800 |
Это значение намного ниже исходного 0,20, поэтому вычисление повторяется с р = 0,15 и q = 0,85. В результате получаем
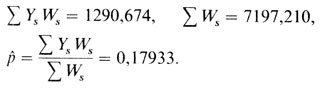
Интерполяция имеет вид
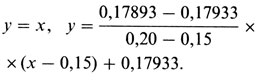
Приравнивая правые части, получаем
Это значение можно использовать для вычисления дисперсии (уравнение П.3.2)

Оценка равна рˆ = 0,1791 ± 0,01224. И это значение отличается от ожидаемого при рецессивном наследовании. Кроме того, известно, что в достаточном количестве случаев признак имеет экзогенную природу. Возможно, авторам не удалось исключить все такие случаи из своих данных. Следовало бы выделить их как спорадические, которые увеличивают число семей только с одним пораженным (табл. П.3.3).
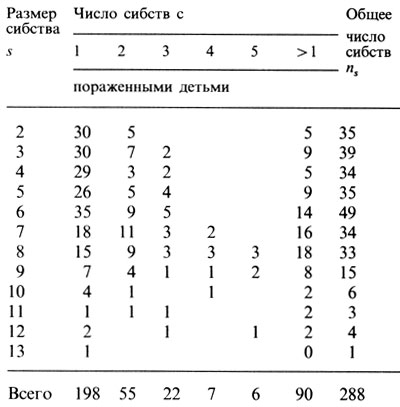
Таблица П.3.3. Ответ на вопрос: чаще ли встречаются сибства с одним пораженным, чем можно ожидать?
Ожидаемое значение этой величины, вычисленное из табл. П.3.3, равно 181,56, а реально наблюдаемое - 198. По формуле П.3.5 дисперсия получается 59,052. Сравнение дает χ2 = (198 - 181,56)/√59,052 = 2,139, Р < 0,05 (односторонний критерий), т. е. спорадических случаев слишком много, поэтому полагаем rmin = 2. Теперь, используя формулы для Bs и Ws из уравнения П.3.1, получаем для k = 0: формулы для rmin = 1, k = 1, где s заменено на s - 1;
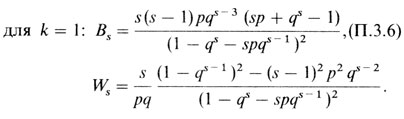
Следовательно, определение р нужно повторить, используя только сибства по крайней мере с двумя пораженными (90 сибств с 234 сибсами). Наша предварительная оценка рˆ составляет 0,25. Расчет дает
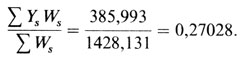
Затем расчет, повторенный для рˆ = 0,30, дает
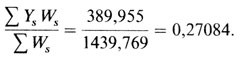
Окончательную оценку получаем путем интерполяции. Стандартное отклонение можно вычислить по формуле (П.3.2):
Полученная теперь величина очень хорошо согласуется с ожидаемым сегрегационным отношением 0,25. На ее основе можно оценить число спорадических случаев, не наследующихся в семьях по крайней мере с двумя пораженными, а только в семьях с одним пораженным.
Из всех n = ∑ns семей в ∑ns,h будут наследуемые случаи. Значение ns,h можно получить из биномиального распределения, используя величину рˆ и число семей по крайней мере с двумя пораженными детьми ns,r>1 по следующим формулам:
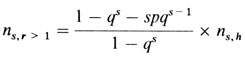
или
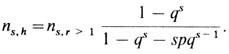
В нашем случае
Конечно, можно спорить, что в этом случае предпочтительнее использование теоретического сегрегационного отношения 0,25. Имеются аргументы как за, так и против этого. Однако различие мало. В случае теоретического сегрегационного отношения было бы получено ∑ns,h = 193,08 семей. На самом деле их оказалось 288. Это означает, что среди спорадических случаев в среднем должно быть 107,97 ненаследственных. Помимо этого имеется 21 случай, для которого нет такой информации, поскольку это одиночные дети. Если мы предположим, что среди них существует та же доля ненаследственных случаев, т. е. 21 × 108/432 = 5,25 случаев, то в сумме получим 113,22 ненаследственных случая (= 24,99%) из 432 + 21 = 453 случаев, где оба родителя непоражены.
Альтернативный способ изучения проблемы ненаследственных случаев заключается в оценке сегрегационного отношения только среди детей из близкородственных браков (табл. П.3.4). В этом случае величина равна рˆ = 0,269 ± 0,038, т. е. совпадает с оценкой, полученной для семей по крайней мере с двумя пораженными детьми.
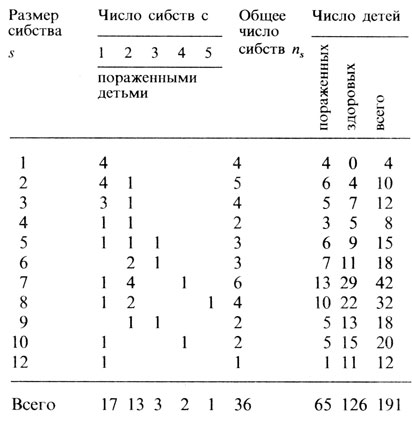
Таблица П.3.4. Сибства из близкородственных браков
До сих пор мы анализировали только брак двух здоровых людей. Исследуем теперь браки между двумя пораженными (табл. П.3.5). Этот тип брака весьма распространен: ассортативное скрещивание предопределяется системой образования, которая создает "социальный изолят" для глухонемых. Если бы глухонемота всегда вызывалась одним и тем же рецессивным геном, то все дети в этих браках были бы лишены слуха. Такая ситуация имеет место только в 5 сибствах, тогда как в 6 браках наблюдаются как пораженные, так и непораженные дети. Помимо этого авторы зарегистрировали не менее 21 брака между глухонемыми партнерами, чьи дети (всего 53) были здоровы. Анализируя эти данные, можно было бы сделать вывод, что существует генетическая гетерогенность, в основе которой лежит ряд разных рецессивных генов. Однако необходимо учитывать и другую возможность: глухонемота может иметь экзогенную природу. Свидетельства этому были обнаружены в семьях с двумя пораженными супругами в 12 случаях и в семьях с одним пораженным супругом в 5 случаях.

Таблица П.3.5. Сибства из браков типа глухонемой × глухонемой по крайней мере с одним пораженным ребенком
Информация, которую можно получить из браков двух пораженных, многообразна. Рассмотрим те из них, в которых имеются как пораженные, так и непораженные дети (не менее 6 браков с 11 здоровыми и 14 больными детьми). Такие браки нельзя объяснить ни генетической гетерогенностью, ни экзогенными факторами. Наиболее очевидное объяснение состоит в том, что помимо рецессивных мутаций, детерминирующих глухонемоту, имеются также доминантные.
Проанализируем теперь третий тип брака: глухонемой × здоровый (табл. П.3.6). Обследовано 45 таких браков по крайней мере с одним ребенком. В 39 из них наблюдались только здоровые дети, общим числом 102. Очевидно, эта ситуация не противоречит гипотезе о том, что пораженный родитель страдает рецессивным типом заболевания. Однако имеется 6 браков по крайней мере с одним пораженным ребенком. В соответствии с уравнениями П.3.1 и П.3.2 для случая k = 1 оценка сегрегационного отношения равна рˆ = 0,548 ± 0,119.

Таблица П.3.6. Сибства из браков типа глухонемой × здоровый по крайней мере с одним пораженным ребенком
По-видимому, такие браки можно трактовать как браки между гетерозиготами и гомозиготами. В этом случае имеются две возможности: либо мутации, обусловливающие патологию - доминантные (в этом случае пораженный родитель гетерозиготен, а непораженный - гомозиготен), либо рецессивные (в этом случае непораженный родитель гетерозиготен, а пораженный - гомозиготен). Имеющиеся 6 браков не позволяют дискриминировать эти две возможности. На первый взгляд кажется невероятным, что 6 из 45 пораженных случайно имели супруга-гетерозиготу. Однако этот аргумент снимается высоким уровнем ассортативности браков в семьях глухонемых, которая, естественно, приводит не только к бракам между пораженными, но также между пораженными и гетерозиготами, например сибсами и другими близкими родственниками глухонемых.
Популяционная генетика глухонемоты. Авторы сравнивали репродуктивные способности больных с таковыми в общей популяции того же возраста (который, кстати, был очень высоким). Доля вступивших в брак и среднее число детей были несколько снижены. Этот результат подтверждает вывод о том, что среди "ненаследственных случаев" могут встретиться лица с новыми доминантными мутациями. Однако их число невозможно оценить даже приблизительно. Кроме того, важно помнить, что существование доминантных типов точно не доказано. Следовательно, оценка уровня соответствующих мутаций, основанная на таких данных, некорректна. Для рецессивных типов также нельзя получить оценки мутационного уровня (разд. 5.1.3.1). Как указано в табл. П.3.5, многие браки между здоровыми, в которых обнаруживаются дети с дефектами слуха, это браки близкородственные. Теоретически такие наблюдения можно использовать для дальнейшего анализа генетической гетерогенности. Пусть q будет частотой рецессивного аллеля, а с - частотой браков двоюродных братьев и сестер. Тогда относительная частота q браков двоюродных братьев и сестер среди родителей гомозигот зависит от q. Она вычисляется по формуле (разд. 6.3.1.2)
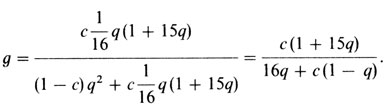
В этой выборке q2 = 0,00027. При с = 0,1% g будет 0,47%, если предположить наличие только одного рецессивного гена. Для с = 1% g равно 4,57%. Реальная доля с для североирландской популяции того же возраста оценивалась авторами величиной 0,1%-0,4%. 6,8% близкородственных браков среди проанализированных семей несовместимы с предположением об одном рецессивном гене, этот результат свидетельствует о генетической гетерогенности.
Однако сам этот аргумент следует рассматривать осторожно. Изоляты внутри популяции могут вызвать тот же эффект. А социальная группа, образуемая глухонемыми и их семьями, обладает многими свойствами социального изолята. Нет необходимости добавлять, что выяснение числа вовлеченных рецессивных генов (хотя бы формальное, если частоты этих генов предполагать идентичными) потребовало бы так много нетестированных и нетестируемых предположений, что такое вычисление просто неоправданно.
Выводы. Рассматриваемая выборка включает все случаи врожденной и рано начавшейся глухонемоты в Северной Ирландии.
На основе клинических обследований несколько случаев были диагностированы как экзогенные и исключены из дальнейшего анализа. Однако клинические данные не позволили выявить всех больных, глухота которых была связана с экзогенными причинами, и их анализировали вместе с больными, имеющими дефект наследственного происхождения.
Соотношение различных типов потомства в браках анализировали статистически. Браки между непораженными дали значимо меньшую оценку сегрегационного отношения р, чем ожидаемое при рецессивном наследовании значение 0,25. Как показал дальнейший анализ, эта более низкая оценка является следствием примеси спорадических случаев. Ограничение статистического анализа сибствами по крайней мере с двумя пораженными детьми или детьми из близкородственных браков дает оценки, согласующиеся с ожидаемыми при аутосомно-рецессивном наследовании. Была оценена доля спорадических случаев во всех сибствах только с одним пораженным ребенком и непораженными родителями. Оказалось, что многие из этих спорадических случаев, очевидно, имеют ненаследственное происхождение, подтверждая клинический опыт, свидетельствующий о роли экзогенных факторов. Однако некоторые из них могут быть следствием доминантных мутаций, особенно потому, что браки глухонемых с непораженными или друг с другом приводили к существенно меньшему количеству случаев, которые наилучшим образом соответствуют критерию доминантного наследования.
Внутри аутосомно-рецессивной группы генетическая гетерогенность подтверждалась тем, что в большинстве браков двух пораженных дети имели нормальный слух. В пользу этого вывода говорило наличие относительно большого числа близкородственных браков среди непораженных родителей глухонемых. По нашему мнению, никаких других выводов из этого исследования сделать нельзя.
Почему так подробно обсуждался этот пример? Чтобы показать, что сегрегационный анализ, использующий рекомендуемые здесь методы, может быть проведен самим исследователем. На регистрацию и обследование семей, как правило, он тратит много месяцев и даже лет. Важно отвести необходимое время и на статистический анализ. Проведение его исследователем, собравшим данные, дает одно большое преимущество. Каждый шаг такого анализа можно оценивать в свете уже собранной информации, клинических результатов и предварительного знания популяции. Никто не "чувствует" данные лучше, чем тот, кто их собирал. Следовательно, для критического осмысления материала больше всего подходят те исследователи, которые его собирали. Конечно, здесь необходим совет статистически образованного коллеги. Сбор материала должен планироваться и проводиться в соответствии с хорошо определенными строгими правилами, от которых нельзя отступать в ходе исследования. Большинство статистических ошибок в генетике человека и вообще в науках о жизни вызваны не тем, что статистические методы неадекватны сами по себе, а тем, что эти методы неправильно используются. Нужно подходить критически к взаимоотношению между сбором материала и его анализом.
Данные по глухонемоте были подвергнуты статистической обработке с использованием сложных методов. Из этого анализа сделаны выводы, касающиеся многих параметров, например частоты доминантных и рецессивных мутаций, количества вовлеченных рецессивных генов. Мы считаем полученные выводы недостаточно обоснованными.
Та же группа исследователей предлагает пакет программ для "сегрегационного анализа", включая оценивание частот, разграничение типов наследования, оценивание числа рецессивных генов и уровня мутаций [800]. Конечно, хорошо иметь данные, проанализированные статистически безупречным способом. Однако необходимо осознавать неизбежные изъяны своего материала и не считать компьютерный анализ панацеей от всех бед.
Генетическая гетерогенность глухонемоты. С момента публикации исследования Стевенсона и Чизмена была проведена большая работа по изучению этого признака. В ходе ее подтвердилась генетическая гетерогенность и существование доминантных типов. Несколько наследственных типов удается теперь идентифицировать на основе клинических и биохимически распознаваемых симптомов [825; 669]. Развитие слуха у человека - сложный процесс, в нем участвует множество генов. Нарушение в любом из них может привести к глухоте.
Поправка на смещения вследствие регистрации в семьях по крайней мере с двумя сибствами и с разными типами пробандов. Часто обследуются семьи, которые содержат более одного сибства. Кроме того, могут существовать разные типы пробандов. Например, семьи с реципрокными транслокациями могут быть зарегистрированы через носителя несбалансированной транслокации, в большинстве случаев ребенка с множественными уродствами. Пробанд может быть носителем сбалансированной транслокации, он может быть зарегистрирован в ходе хромосомного скрининга, такого, например, который проводится во взрослых нормальных популяциях, в популяциях новорожденных, среди лиц с задержкой умственного развития, среди любых индивидов, обладающих определенными уродствами, или при изучении спонтанных абортов. Семьи, зарегистрированные по спонтанным абортам, будут исследованы корректно, только если произошло по крайней мере два аборта. Кроме того, результаты будут зависеть от того, основывался ли анализ на данных одного автора или они получены в ходе совместного исследования. Последний случай предпочтителен, поскольку существует меньшая опасность комбинации "интересных случаев". Шафер [501а] в своем исследовании по сегрегации транслокаций обсуждал эти проблемы и высказал предложение относительно поправок для наиболее важных смещений.
При сборе семейных данных обязательно должен определяться тип регистрации. Большинство опубликованных случаев обычно зарегистрировано через ребенка с несбалансированной транслокацией. На первый взгляд, по-видимому, подходит статистическая коррекция в соответствии с моделью единичного отбора (k = 0) в сибстве этого ребенка-пробанда. Можно спорить о том, что регистрация таких семей зависит от клинического статуса других родственников, поэтому разумно повторить вычисления, используя модель усеченного отбора. Истинное сегрегационное отношение может оказаться ближе к результату, полученному на основе модели единичного отбора. Однако это справедливо только в том случае, если анализ основывается на семьях, зарегистрированных через клинически пораженного индивида. В будущем все больше и больше семейных исследований будут включать длительное и полное наблюдение всех новорожденных с врожденными пороками в целой популяции, как это уже делается, например, в Венгрии [616; 617]. В таких случаях адекватна модель усеченного отбора.
По-видимому, эти родословные будут состоять по крайней мере из двух сибств и характеризоваться наличием дополнительных (вторичных или даже третичных) пробандов. Все индивиды, побудившие исследователя расширить свои изыскания и обследовать другие поколения или сибства, должны рассматриваться в качестве пробандов. Объясним это на примере модельной родословной (рис. П.3.1, табл. П.3.7 [501а]).
![Рис. П.3.1. Модельная родословная с транслокациями. Анализ приведен в табл. П.3.7 [501а]](pic/000153.jpg)
Рис. П.3.1. Модельная родословная с транслокациями. Анализ приведен в табл. П.3.7 [501а]
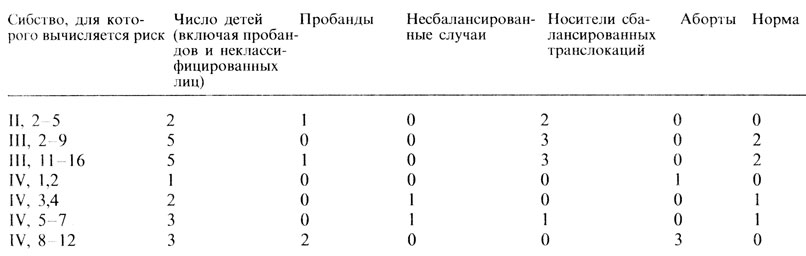
Таблица П.3.7. Данные для вычисления риска в семьях (см. рис. П.3.1)
Семья зарегистрирована через носительницу III,16. Она была кариотипирована, поскольку страдала невынашиванием беременности. Процедуру кариотипирования обычно проводят для женщин, у которых произошло два спонтанных аборта; два абортуса рассматриваются в качестве пробандов среди ее детей и исключаются из процедуры вычисления риска для абортусов. III,16 является "вторичным пробандом". Кроме того, если бы ее мать (II,5) имела нормальный кариотип, то III,16 можно было бы считать носительницей транслокации de novo и сибство, к которому принадлежит II,5, не нужно было бы обследовать. Следовательно, II,5 является другим вторичным пробандом и должна быть исключена из процедуры вычисления риска для ее сибства. II,4 не была кариотипирована. Поскольку у нее нет детей, то неизвестно, является ли она носителем или нет. Следовательно, и ее следует исключить из процедуры вычисления риска. Будучи сестрой пробанда, II,11 должна быть обследована в любом случае, независимо от наличия детей. Следовательно, она не учитывается в качестве пробанда и должна быть учтена в процедуре вычисления риска для детей носителей транслокации. У нее трое детей, которых необходимо обследовать в любом случае, независимо от их фенотипов. Следовательно, поправка не нужна: они могут быть включены в процедуру вычисления риска.
Следует тщательно провести следующий этап анализа. Поскольку представителей II поколения обследовали в любом случае, 11,5 является единственным пробандом в этом поколении, а всех других сибсов можно использовать в процедуре вычисления риска (исключая, конечно, II,4). Кроме того, сибства III,2...9, IV,1,2 и IV,3,4 были зарегистрированы через пораженного родителя, поэтому коррекция не нужна. Однако если, например, сибство IV,3,4 было обследовано лишь потому, что III,11 сообщила исследователю, что у ее двоюродного брата тоже есть ребенок с врожденными пороками (и если это сибство иначе не было бы обследовано), то ребенок IV,4 с несбалансированной транслокацией является (третичным) пробандом и должен быть исключен из процедуры оценивания риска. Этот пример показывает, насколько важно точное и полное описание процесса регистрации. Далее предполагается, что сибства в левой части рис. П.3.1 на самом деле зарегистрированы через пораженного родителя. Результат приведен в табл. П.3.7. Получены следующие оценки риска:
а) для больных с несбалансированной 2/21, транслокацией
б) для абортусов 4/21,
в) для носителей сбалансированных 9/21, транслокаций
г) для нормальных детей 6/21.
Для получения этих оценок были просто объединены единичные случаи из всех сибств (= предварительное накопление). Эту процедуру можно подвергнуть критике на том основании, что сибства большего размера имеют намного больший вес, чем сибства меньшего размера. Можно, конечно, получить оценки риска для каждого сибства отдельно, а уже затем их объединить (= последующее накопление). Однако большинство исследований по транслокациям было выполнено с использованием предварительного накопления. Эта процедура оказывается оправданной, если сибства принадлежат родословным большого размера, поскольку можно предполагать, что в такой родословной реальные риски будут одинаковыми во всех сибствах. С другой стороны, такие вычисления риска необходимо провести отдельно 1) для семей, которые были зарегистрированы через абортусов и 2) которые были зарегистрированы через носителей сбалансированных транслокаций, поскольку лишь у некоторых из обладателей несбалансированных транслокаций могут родиться дети также с несбалансированными транслокациями (лишь немногие зиготы с несбалансированной транслокацией способны развиваться). По существу те же правила вычисления риска следует применить к большим родословным с аутосомно-доминантными или Х-сцепленными болезнями.
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'