2.2. Хромосомные заболевания человека
2.2.1. Синдромы, связанные с аномалиями числа хромосом
Механизмы, лежащие в основе геномных мутаций (аномалии числа хромосом). Аномалии числа хромосом могут быть вызваны разными причинами:
- Наиболее важным механизмом является нерасхождение. Хромосомы, которые в норме должны разделиться во время клеточного деления, остаются соединенными вместе и в анафазе отходят к одному полюсу. Это может произойти в ходе митотического деления, но чаще наблюдается во время мейоза. У человека по неизвестным причинам именно акроцентрические хромосомы имеют тенденцию чаще вовлекаться в нерасхождение (разд. 5.1.2). Мейотическое нерасхождение было открыто Бриджесом (1916) [311] у дрозофилы. На каждую гамету с одной добавочной хромосомой приходится другая, без одной хромосомы. После оплодотворения гаметой с нормальным набором хромосом зигота оказывается по одной из хромосом либо трисомной, либо моносомной. Соматическое нерасхождение в митотически делящихся клетках во время раннего развития может приводить к мозаицизму с наличием нормальных клеток, трисомиков и моносомиков.
- Вторым механизмом, обусловливающим геномные мутации, является утрата отдельной хромосомы вследствие "анафазного отставания": во время анафазного движения одна хромосома может отстать от всех других. Утрата хромосом ведет к мозаицизму, при котором имеются одна эуплоидная и одна моносомная клеточная популяция. У мыши стадия пронуклеусов (т. е. период между проникновением ядра спермия в ооцит и слиянием двух гаплоидных родительских ядер) особенно чувствительна к утрате отцовской X-хромосомы. Этот период, как и первые стадии дробления, вероятно, весьма чувствителен и у человека, поскольку многие мозаики формируются именно на этой стадии (разд. 5.1.6).
- Третьим механизмом является полиплоидизация. При этом в каждой клетке геном целиком представлен более чем дважды. У человека обнаружена только триплоидия, при которой число хромосом равно 3n=69.
Аномальное число хромосом в клетке (анеуплоидия) увеличивает риск последующих нарушений, таких, как потеря хромосом вследствие анафазного отставания в последующих клеточных делениях. Для многих случаев мозаицизма с двумя клеточными популяциями, состоящими из равных пропорций трисомных и эуплоидных клеток, такое объяснение представляется наиболее удовлетворительным (разд. 5.1.6). Хромосома, лишенная партнера, в таких случаях, по-видимому, мешает нормальной конъюгации двух других гомологов.
Синдром Дауна. Это наиболее частое хромосомное заболевание человека. Его частота среди новорожденных 1-2/1000, и именно этот синдром является наиболее распространенной причиной обращения в медико-генетические консультации. Рис. 2.27 показывает, что физические различия между тремя основными расовыми группами существенно перекрываются фенотипическим сходством всех больных. На рис. 2.28 приведены наиболее частые клинические симптомы. Наиболее важными характеристиками синдрома являются следующие:
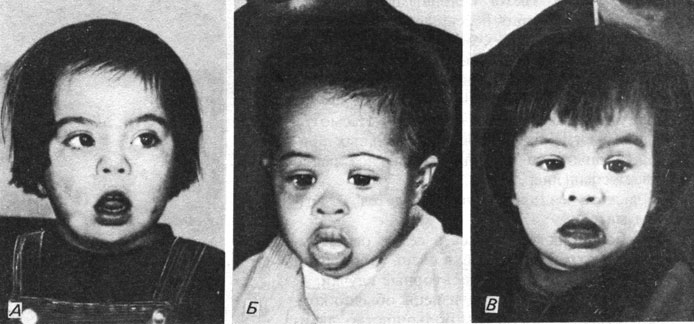
Рис. 2.27. Дети с синдромом Дауна. А. Европеоид. Б. Негр. В. Представитель азиатской расы. Общие признаки синдрома Дауна более заметны, чем расовые различия. (Courtesy of Dr. T. M. Schroeder-Kurth.)
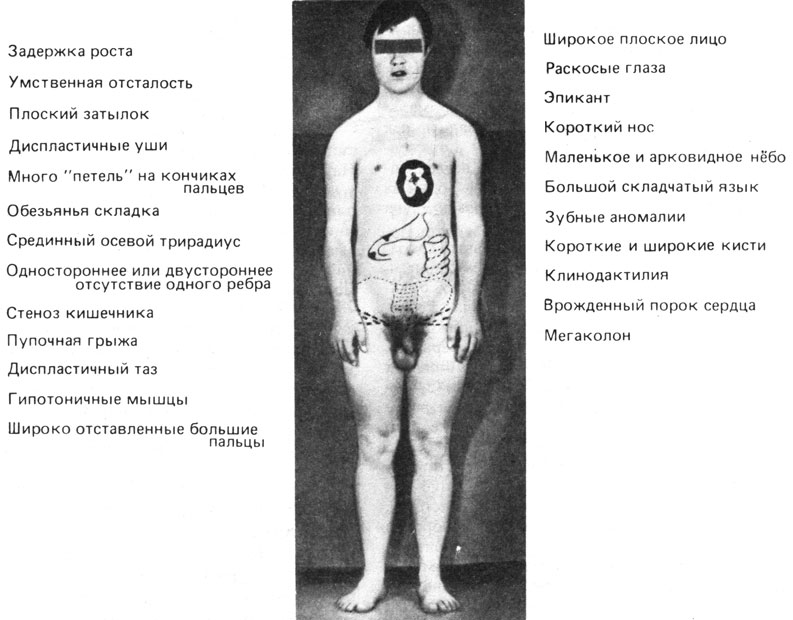
Рис. 2.28. Основные клинические симптомы болезни Дауна
а) это четко очерченное состояние. Несмотря на значительную изменчивость отдельных признаков, у опытного клинициста диагноз редко вызывает сомнение;
б) частота синдрома увеличивается с возрастом матери;
в) в большинстве случаев в семье регистрируется только один больной; в очень небольшом числе семей наблюдаются повторные случаи;
г) монозиготные (МЗ) близнецы обычно конкордантны, в то время как большинство дизиготных близнецов дискордантны. Из этого правила, однако, есть исключения - иногда встречаются дискордантные пары МЗ [545]. Это связано, вероятно, с утерей лишней хромосомы той клеткой, из которой сформировался нормальный партнер;
д) мужчины с синдромом Дауна бесплодны, однако описано по крайней мере 17 женщин с этим синдромом, у которых были дети. Среди 19 таких детей (включая одну пару МЗ близнецов) у 7 имеется синдром Дауна, 9 - нормальные, 2 - умственно отсталые без синдрома Дауна и 2 мертворожденных МЗ близнеца - с нормальными кариотипами, которые учитывались как один индивид [443]. Все матери и пораженные дети, у которых было проведено исследование хромосом, имели кариотип 47,G +, один из умственно отсталых детей без синдрома Дауна имел нормальный кариотип 46, XY;
е) продолжительность жизни больных сокращена [160]. Согласно австралийским данным, опубликованным еще в 1963 г. [327], 31,1% больных умирают в конце первого года жизни, 46% - в конце третьего года. Продолжительность жизни укорочена и в поздних периодах жизни. В другой выборке [465] 37 из 73 больных умерли от респираторных заболеваний (туберкулез не учитывался), что в 123 раза выше частоты смертельных случаев по тем же причинам в общей популяции того же возраста. 5 больных умерли от других инфекций. Эти данные дают основания предполагать наличие при болезни Дауна дефекта иммунной системы. Увеличена также частота врожденных пороков сердца. С появлением антибиотиков и развитием сердечной хирургии эти больные живут намного дольше (рис. 2.29). Однако вряд ли крайние значения продолжительности жизни будут слишком большими, поскольку предполагается, что больные с синдромом Дауна стареют быстрее, чем нормальные люди;
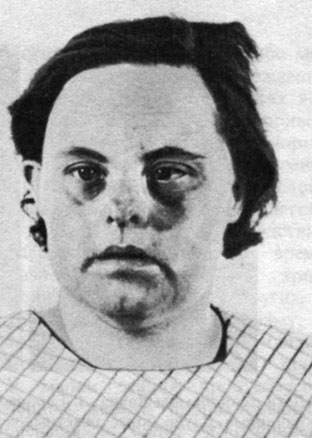
Рис. 2.29. Женщина с синдромом Дауна в возрасте 38 лет. (Courtesy of Dr. T. M. Schroeder-Kurth.)
ж) степень выраженности отдельных фенотипичёских характеристик синдрома изменчива. Например, врожденный порок сердца отмечается у некоторых, но не у всех больных, и это верно для многих других клинических признаков, описанных выше и перечисленных на рис. 2.28. Такая высокая изменчивость фенотипических проявлений характерна для всех хромосомных синдромов человека;
з) в 20 раз повышен риск смерти от острого лейкоза. Причины этого неизвестны. Существует три гипотезы: высокий риск анеуплоидии, связанный с митотическими нарушениями в стволовых клетках крови, сниженная резистентность к инфекции лейкозогенными вирусами и, как показывают экспериментальные данные, низкая эффективность системы репарации (разд. 5.1.16).
Стандартный кариотип при синдроме Дауна. Хромосомы группы G больного с синдромом Дауна представлены на рис. 2.30 (окраска G- и Q-методом). По рисунку сегментации хромосомы 21 и 22 легкоразличимы, хромосома 21 имеет более сильно флуоресцирующий широкий сегмент и один или два темных G-сегмента. Хромосома 22 имеет темный G-сегмент в проксимальной части длинного плеча и слабоокрашенный - более дистально. В течение короткого периода после открытия трисомии 21 были описаны отдельные случаи болезни Дауна якобы без добавочной хромосомы 21. Однако теперь общепризнано, что каждый больной с этим синдромом имеет дополнительную хромосому либо в форме регулярной трисомии 21, либо в форме транслокации, образованной хромосомой 21 и другой хромосомой (чаще всего 21, 22, 13, 14, 15). Наблюдения редких случаев реципрокной транслокации позволяют сделать вывод, что именно дистальный район длинного плеча хромосомы 21, в частности сегмент 21q22, ответствен в случае его трисомии за возникновение характерного фенотипа [371]. Например, у девочки, кариотип которой помимо одной нормальной хромосомы 21-й пары содержит дуплицированный второй гомолог 21, но без сегмента 21q22 (рис. 2.31), отмечалась умеренная умственная отсталость, но у нее отсутствовала большая часть признаков синдрома Дауна. В то же время трисомия только по одному сегменту 21q22 приводит к мягким проявлениям этого синдрома [370].
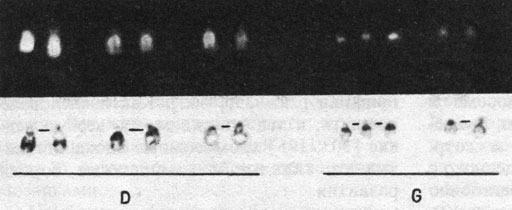
Рис. 2.30. D- и G-хромосомы больного с синдромом Дауна. Окрашивание Q- и G-методом. Обратите внимание на широкий сегмент в проксимальном районе 21p, по которому хромосома 21 отличается от хромосомы 22. (Courtesy of Dr.T.M. Schroeder-Kurth.)
![Рис. 2.31. А. Тандемная дупликация в одной из хромосом 21-й пары, не захватывающая сегмент 21 q 22 (средняя хромосома), у ребенка с неглубокой умственной отсталостью и с отсутствием многих признаков синдрома Дауна [371]. Б. Аномальная хромосома (слева) и для сравнения две нормальные хромосомы 21, совмещенные на фотографии теломерами (справа). Здесь в отличие от хромосомы с дупликацией полоса 21 q 22 выглядит удвоенной](pic/000037.jpg)
Рис. 2.31. А. Тандемная дупликация в одной из хромосом 21-й пары, не захватывающая сегмент 21 q 22 (средняя хромосома), у ребенка с неглубокой умственной отсталостью и с отсутствием многих признаков синдрома Дауна [371]. Б. Аномальная хромосома (слева) и для сравнения две нормальные хромосомы 21, совмещенные на фотографии теломерами (справа). Здесь в отличие от хромосомы с дупликацией полоса 21 q 22 выглядит удвоенной
Синдром Дауна был известен как клинически самостоятельное заболевание задолго до трисомии 21. Другие синдромы, связанные с аномалиями аутосом, были скрыты среди огромного количества множественных пороков развития, и возможность их выделения как самостоятельных клинических единиц появилась только в результате развития методов хромосомной диагностики. Однако теперь, оценивая ситуацию ретроспективно, можно отметить, что некоторые синдромы настолько своеобразны, что их, вероятно, можно было бы выделить на чисто клинической основе.
Другие аутосомные трисомии. Патау и сотр. (1960) [472] впервые описали случай аутосомной трисомии, отличный от трисомии 21. Это открытие было результатом целенаправленного поиска на основе гипотезы, которая была сформулирована авторами следующим образом:
"С генетической точки зрения маловероятно, что добавление к нормальному набору какой-то аутосомы будет иметь такой же ограниченный эффект, как X-трисомии. В настоящее время известен только один тип аутосомной трисомии, и, хотя лишняя хромосома является одной из двух самых маленьких аутосом, ее наличие в триплицированном состоянии приводит к монголизму... Следует ожидать, что другие аутосомные трисомии, если они совместимы с жизнью, должны также приводить к множественным врожденным порокам".
Благодаря систематическому обследованию новорожденных с множественными пороками развития Патау с сотр. удалось выявить три случая трисомии: двух больных с трисомией по 18-й хромосоме и одного-с трисомией по одной из D-xpoмосом. Одновременно Эдварде и сотр. [343] также обнаружили новорожденного с трисомией 18 (первоначально ошибочно идентифицированную как трисомия 17). Трисомия D позже была идентифицирована как трисомия 13. Основные признаки и симптомы заболеваний, связанных с этими хромосомными аномалиями, представлены на рис. 2.32 и 2.33. В последующие годы все попытки открыть новые синдромы аутосомных трисомии среди новорожденных оказались безуспешными, на основании чего был сделан вывод о том, что они летальны. Этот вывод был подтвержден исследованиями хромосом при спонтанных абортах; в клетках таких эмбрионов обнаруживались и другие варианты трисомии. Открытие трех новых синдромов - трисомии 8, 9 и 22 - последовало после разработки методов дифференциального окрашивания [292, 396, 402]. Как и следовало ожидать, и эти, по-видимому, весьма редкие [503, 195] хромосомные аномалии вызывают тяжелые и комплексные пороки развития.
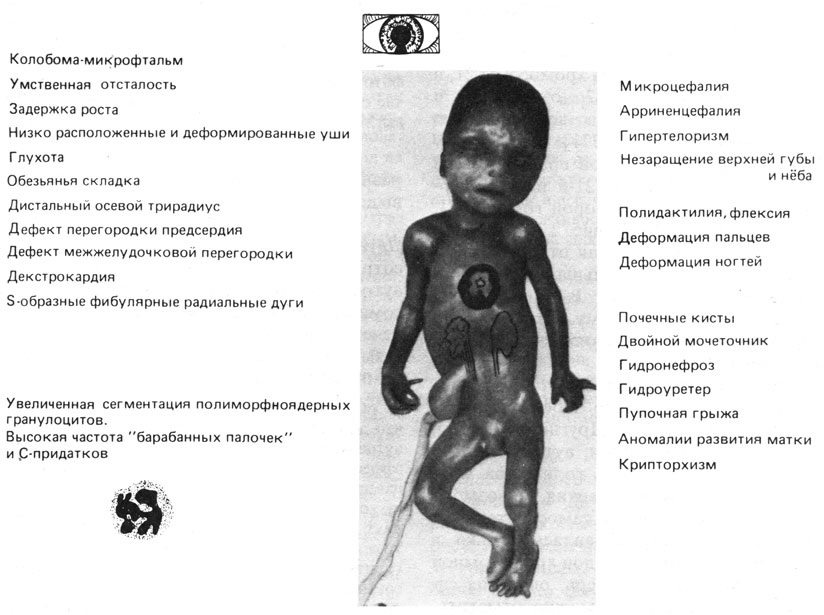
Рис. 2.32. Основные клинические симптомы трисомии по хромосоме 13
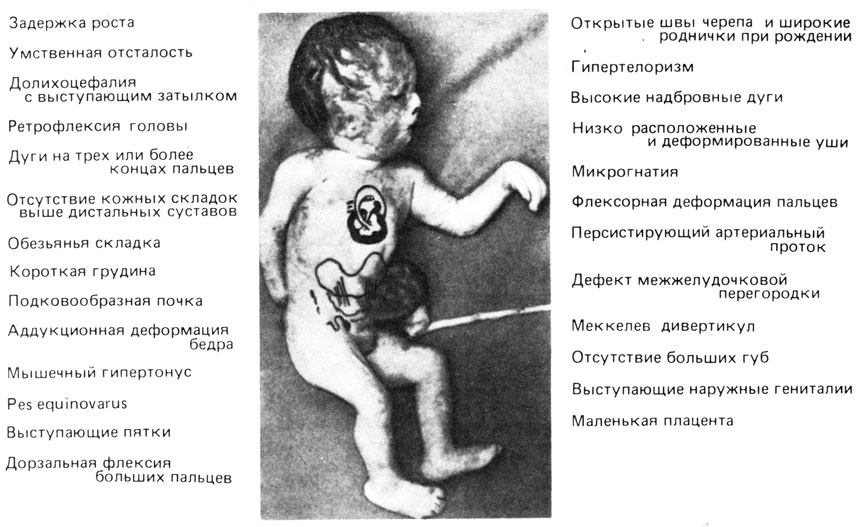
Рис. 2.33. Основные клинические симптомы трисомии по хромосоме 18
Триплоидия. Первые примеры триплоидии обнаружены у двух абортированных плодов [335, 475]. Приблизительно в это же время описан сомнительный случай мозаицизма [308]. Более поздние исследования показали, что триплоидия у спонтанных абортусов не так уже редка, а в очень небольшом числе случаев наблюдается даже у живорожденных детей [458]. К 1974 г. на основании изучения 275 триплоидных абортусов, полученных при сроках беременности менее чем 20 недель, была накоплена более или менее детальная информация. Двадцать два из исследованных плодов достигли возраста 28 недель; пять других погибли in utero; остальные прожили несколько часов или дней после рождения. Все живорожденные дети, прожившие дольше нескольких дней (к 1974 г. их было 8), оказались триплоид-диплоидными мозаиками.
Наиболее характерным признаком триплоидии является пузырное перерождение плаценты (mole hydatidiforme). У некоторых эмбрионов обнаруживаются локальные пороки развития, но часть плодов имеет как будто бы нормальный фенотип.
Триплоиды, родившиеся живыми, имеют небольшой вес, широкий задний родничок с недоразвитыми затылочными и теменными костями черепа и другие неспецифические аномалии, которые характерны для многих аутосомных аберраций. Триплоиды мужского пола с кариотипом 69, XXY характеризуются нарушением гениталий: у них маленький половой член в сочетании с гипоспадией, расщепленной мошонкой и неопустившимися яичками. Некоторые из мозаиков выживают. Клинические признаки не очень четкие, предварительный диагноз можно поставить на основании умственной отсталости в сочетании с аномалиями плаценты, синдактилией, аномалиями гениталий и асимметрией.
Триплоидия возникает вследствие ошибок при образовании половых клеток (рис. 2.34). Различия в причинах появления триплоидов определяют среди них соотношения индивидов с генотипами XXX, XXY и XYY. Существуют факты, свидетельствующие о том, что причиной триплоидии может быть двойное оплодотворение или отсутствие первого мейотического деления ооцита [1504, 414].
![Рис. 2.34. Аномалии оогенеза, сперматогенеза или оплодотворения, обусловливающие триплоидию. А. Мужчина может оказаться триплоидным, если у его отца имелся тетраплоидный сперматогоний или был нарушен мейоз. Триплоидия у мужчин может быть и результатом оплодотворения двумя сперматозоидами (По Niebuhr, Hum Genet., 21, 1974). Б. У женщин, так же как у мужчин, триплоидия может объясняться нарушением гаметогенеза в предыдущем поколении. В. Аномальное деление зиготы или клеток эмбриона приводит к мозаицизму [458]](pic/000040.jpg)
Рис. 2.34. Аномалии оогенеза, сперматогенеза или оплодотворения, обусловливающие триплоидию. А. Мужчина может оказаться триплоидным, если у его отца имелся тетраплоидный сперматогоний или был нарушен мейоз. Триплоидия у мужчин может быть и результатом оплодотворения двумя сперматозоидами (По Niebuhr, Hum Genet., 21, 1974). Б. У женщин, так же как у мужчин, триплоидия может объясняться нарушением гаметогенеза в предыдущем поколении. В. Аномальное деление зиготы или клеток эмбриона приводит к мозаицизму [458]
Мозаики. Мозаиками называют особей, в организме которых сосуществуют две или более генетически различных клеточных популяции. Мозаицизм обнаруживается довольно часто при численных аномалиях как половых хромосом, так и аутосом. Хромосомных мозаиков иногда называют миксоплоидами. Мозаик может возникнуть вследствие митотического нерасхождения или в результате утери хромосомы вследствие анафазного отставания (рис. 2.35). Оценки частоты таких нарушений митоза получены в случае синдрома Дауна. Риск анафазного отставания в 400 раз выше в трисомной зиготе, чем в эуплоидной, а митотического нерасхождения - в 70 раз. Эти оценки основаны на сопоставлении относительных частот различных типов мозаицизма (разд. 5.1.6) и на анализе эффекта возраста матери. Частота мозаиков, возникающих вследствие мейотического нерасхождения с последующей утратой дополнительной хромосомы в анафазе теоретически должна увеличиваться с возрастом матери, так же, как и при обычных гаметических трисомиях. В то же время частота мозаиков, возникших в результате нерасхождения хромосом в митозе, не должна зависеть от возраста матери. Следовательно, долю мозаиков, возникших вследствие анафазного отставания, можно оценить при сравнении эффекта возраста матери на частоту мозаицизма и гаметических трисомий. Однако точную оценку получить трудно, поскольку некоторые мозаики не диагностируются: при ограниченном числе клеток, используемых для кариотипирования, аберрантные можно пропустить, т. к. их очень мало. Кроме того, мозаики с небольшим количеством аберрантных клеток характеризуются соответственно и невыраженными фенотипическими отклонениями (если таковые вообще есть). Такие мозаики обнаруживаются случайно, главным образом когда трисомные клетки имеются в их герминативной ткани и в потомстве встречаются трисомики. К настоящему времени среди описанных в литературе случаев мозаицизма 17-30% приходится на митотическое нерасхождение. Как и ожидалось, возраст матери был особенно низким в тех случаях, где доля трисомных клеток (в расчете на все исследованные) составляла менее одной трети [483]. Суммарная частота мозаиков среди всех с клиническими симптомами болезни Дауна составляет приблизительно 2%.
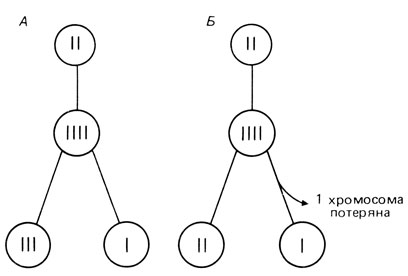
Рис. 2.35. Нерасхождение в митозе (А) и отставание в анафазе (Б). После того как гомологичные хромосомы удваиваются, три из образовавшихся четырех хроматид оказываются в одном продукте деления в следующей анафазе. Б. Одна хромосома отстает во время анафазного движения
Статистические проблемы выявления мозаицизма. Насколько широко распространен хромосомный мозаицизм в популяции по сравнению с другими хромосомными аберрациями? Решение этого вопроса связано с анализом статистических проблем, суть которых состоит в оценке вероятности обнаружения мозаика в зависимости от доли аберрантных клеток в исследуемой ткани и от числа исследованных клеток в выборке. В большинстве опубликованных обзоров (разд. 5.1.2) обычно приводятся данные по небольшому (от 3 до 5) числу клеток одного индивида, в связи с чем доля мозаиков систематически занижается. Важно учесть, что в процессе получения препаратов какие-то хромосомы могут быть утрачены, т. е. возможны артефакты. Отметим также и то обстоятельство, что за короткое время трудно исследовать несколько сотен клеток от одного индивида. В работе Бочкова и сотр. (1974) [306] был предложен пригодный для практики компромисс.
Вначале принимаем допустимый предел для доли аберрантных клеток. Затем общее число клеток, необходимых для обнаружения с вероятностью 95% по крайней мере одной аберрантной клетки, определяется на основе биномиального закона. Так, вероятность того, что среди n проанализированных клеток не встретится ни одной аномальной клетки, составляет (1-p)n, где p - допустимый предел для доли аномальных клеток. Разумно выбрать 25% как нижний предел, пригодный для диагноза мозаицизма, поскольку индивиды, имеющие менее 25% аномальных клеток, обычно характеризуются слабовыраженными клиническими проявлениями. Теперь допустим, что P1,n - это вероятность обнаружения по крайней мере одной аномальной клетки в выборке из n клеток: p=0,25, тогда
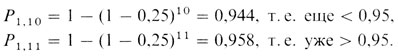
Следовательно, число клеток, которое следует проанализировать, равно 11. Если анеуплоидная клетка не найдена, диагностируется отсутствие мозаицизма или, точнее, утверждается, что имеется не более чем 25% аномальных клеток. Если обнаружена более чем одна клетка с одной и той же аномалией, диагноз мозаицизма подтверждается. При наличии одной аномальной клетки это может быть проявлением мозаицизма или артефактом. Следовательно, размер выборки должен быть увеличен до таких размеров, чтобы обнаружить по крайней мере две аномальные клетки с P2,n=0,95, P2,17=0,951. Соответственно на втором этапе необходимо проанализировать еще шесть клеток в дополнение к первоначальным 11. Если не найдено второй аномальной клетки с такой же аберрацией, первая должна рассматриваться как артефакт. Если найдена вторая клетка, третью следует искать в выборке объемом в 23 клетки и т. д. Усовершенствованный метод предложен Хуком (Amer. J. Hum. Genet. 29, 94-97, 1977).
В клинической практике часто исследуют значительно большее число клеток, так как мозаицизм должен быть исключен с большой достоверностью, а также потому, что в некоторых случаях желательно обнаружить мозаицизм с очень малой пропорцией аномальных клеток.
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'