2.2.2. Синдромы, связанные со структурными аномалиями аутосом
2.2.2.1. Кариотипы и клинические синдромы
Первые наблюдения синдрома Дауна. Как только трисомия 21 была идентифицирована как причина синдрома Дауна, естественно возник вопрос о том, у всех ли больных имеется эта трисомия. Если не у всех, то исключения могли бы представлять большой интерес. Так как риск мейотического нерасхождения, как уже тогда было известно, увеличивается с возрастом матери и поскольку единичное нерасхождение должно вести к появлению только одного пораженного потомка, исключения следовало искать среди пораженных детей молодых матерей, а также в семьях с двумя или более больными.
Полани и сотр. (1960) [479] исследовали трех таких больных с синдромом Дауна. У одной девочки, первого ребенка 21-летней матери и 23-летнего отца, они обнаружили 46 хромосом. Было найдено четыре хромосомы группы G. Однако одна хромосома из группы D имела удлиненное короткое плечо. Авторы предположили, что дополнительная хромосома 21 была транслоцирована на короткое плечо D-хромосомы. Очень скоро это предположение подтвердилось при исследовании семейных случаев. Две здоровые матери трех больных с синдромом Дауна и их общая бабка имели только 45 хромосом и только 3 стандартные хромосомы группы G. Однако одна из хромосом группы D (исследователи предположили, что это хромосома 15) имела удлиненное короткое плечо. Если это плечо содержит материал отсутствующей хромосомы 21, тогда кариотип этих женщин является сбалансированным, т. е. весь генетический материал диплоидного набора присутствует. С другой стороны, у некоторых из их потомков имеется хромосома с транслокацией, включающей большую часть материала хромосомы 21. Фактически у таких детей имеется трисомия 21 и возникает синдром Дауна, несмотря на то что формально число хромосом у них стандартное. Такой кариотип является несбалансированным. Примерно в то же время была описана первая транслокация G/G [354]. Вскоре после этого при исследовании первого мейотического деления у гетерозиготного носителя сбалансированной транслокации был обнаружен тривалент, т. е. фигура, состоящая из трех хромосом, и это послужило четким доказательством того, что нестандартная хромосома, обнаруженная в этих семьях, действительно несет транслокацию [373].
Частота транслокационного синдрома Дауна. Транслокация при синдроме Дауна объясняет много семейных случаев, но не все. Стандартная трисомия 21 может повторно возникать в одной и той же семье, указывая на наличие у родителей каких-то конституциональных факторов, предрасполагающих к нерасхождению, или мозаицизму (разд. 5.1.2). В табл. 2.3 приведены данные о частоте транслокационных случаев (наследуемых и спорадических) среди больных с синдромом Дауна для двух групп матерей: молодых и пожилых. Большинство случаев характеризуется описанными выше транслокациями D/G и G/G.
![Таблица 2.3. Частота транслокаций у детей с синдромом Дауна [443]](pic/000043.jpg)
Таблица 2.3. Частота транслокаций у детей с синдромом Дауна [443]
Существует, однако, небольшое число реципрокных транслокаций, в которые вовлекаются другие - неакроцентрические хромосомы. Детальное обсуждение различных структурных аберраций целесообразно предварить замечаниями относительно механизмов их образования.
Пробелы и разрывы. Необходимым условием возникновения структурной хромосомной перестройки любого типа является наличие в хромосоме разрыва. Если исходить из того, что ДНК представляет собой единую длинную нить, проходящую через всю хромосому, хромосомный разрыв предполагает и разрыв сахаро-фосфатного остова ДНК. В световом микроскопе бывает трудно отличить хромосомный разрыв от ахроматической (неокрашенной) области, называемой пробелом. Эти пробелы могут отражать как истинные разрывы, так и участки локальной деспирализации. Хромосомные разрывы часто учитывают при оценке мутационного процесса, поэтому необходимо прийти к соглашению относительно того, какие аберрации учитывать как разрывы, а какие - как пробелы. Схема, положенная в основу одного из таких соглашений, представлена на рис. 2.36. Указанные в ней отличительные признаки достаточно строгие и, вероятно, занижают количество разрывов. Разрывы и пробелы могут возникать во время интерфазы как до, так и после репликации ДНК. Если разрыв происходит до репликации, повреждение будет видно в последующей метафазе в обеих хроматидах (изохроматидный разрыв). Если событие произойдет после фазы репликации, поврежденной окажется только одна хроматида (хроматидный разрыв). Различные типы разрывов и пробелов представлены на рис. 2.37.
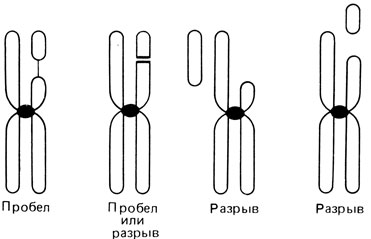
Рис. 2.36. Определение хромосомного пробела и разрыва. Пробел - отдельные сегменты не смещены, между ними может даже сохраниться связь. Если эта связь отсутствует, трудно решить, с чем мы имеем дело, с разрывом или пробелом. Две правые хромосомы демонстрируют разрывы с разной локализацией отделившихся сегментов. (Courtesy of Dr.T.M. Schroeder-Kurth.)
![Рис. 2.37. Различные типы хромосомных пробелов и разрывов. 1. Хроматидные пробелы. 2. Изохроматидные пробелы. 3. Хроматидные разрывы. 4. Изохроматидные разрывы. 5. Микрофрагменты одиночные (А), двойные (Б) [212]](pic/000045.jpg)
Рис. 2.37. Различные типы хромосомных пробелов и разрывов. 1. Хроматидные пробелы. 2. Изохроматидные пробелы. 3. Хроматидные разрывы. 4. Изохроматидные разрывы. 5. Микрофрагменты одиночные (А), двойные (Б) [212]
Судьба поврежденных хромосом. Разрыв, происходящий в любом районе хромосомы и не затрагивающий центромеры, приводит к появлению укороченной хромосомы с центромерой и ацентрического фрагмента. Такой фрагмент иногда может формировать маленькое кольцо, но, будучи лишенным центромеры, чаще всего теряется в последующем митозе. Таким образом, разрыв хромосомы часто приводит к появлению клетки, лишенной хромосомного сегмента. В некоторых случаях, однако, целостность хромосомы, имеющей разрывы в двух точках, восстанавливается ферментами репарации. Механизмы такого воссоединения концов в настоящее время известны [456]. Если концы хромосомных фрагментов воссоединятся друг с другом удачно, то и хромосома, и клетка будут снова интактными. Действительно, исследования при заболеваниях, связанных с недостаточностью репаративных ферментов, показывают, что подобные события могут происходить многократно во многих тканях. В других случаях концы хромосомных фрагментов могут воссоединиться в точках разрыва других хромосом как гомологичных, так и негомологичных (при условии, что два разрыва происходят в пределах относительно короткого отрезка времени и достаточно близко друг от друга). Это приводит к образованию хромосомных перестроек различного типа.
Внутрихромосомные перестройки (внутренние обмены). В пределах одной хромосомы могут произойти разрывы в двух разных участках, и фрагмент между точками разрыва, перевернувшись, может вновь соединиться с хромосомой. Такая перестройка (инверсия) не приводит к нарушениям в митозе, особенно если разрыв произошел в фазе G1. Она может быть обнаружена методами дифференциального окрашивания. В тех случаях, когда инверсия не затрагивает центромеру, она называется парацентрической, если же точки разрыва находятся по обе стороны от центромеры, такую инверсию называют перицентрической. Гетерозиготы по инверсиям не очень редки в популяциях человека (рис. 2.38). Инверсии могут создавать затруднения в конъюгации гомологичных хромосом в мейозе и приводить к частичной элиминации некоторых типов половых клеток у гетерозигот по инверсиям (рис. 2.39). У гомозигот таких затруднений нет. Инверсии (особенно перицентрические), несомненно, играли важную роль в филогении высших приматов (разд. 7.2.1).
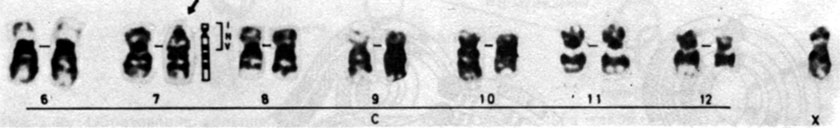
Рис. 2.38. Перицентрическая инверсия хромосомы 7 у здорового мужчины, G-окрашивание. Инвертированный сегмент изображен схематически (INV). Другие хромосомы показаны для сравнения. (Courtesy of Dr.T.M. Schroeder-Kurth.)
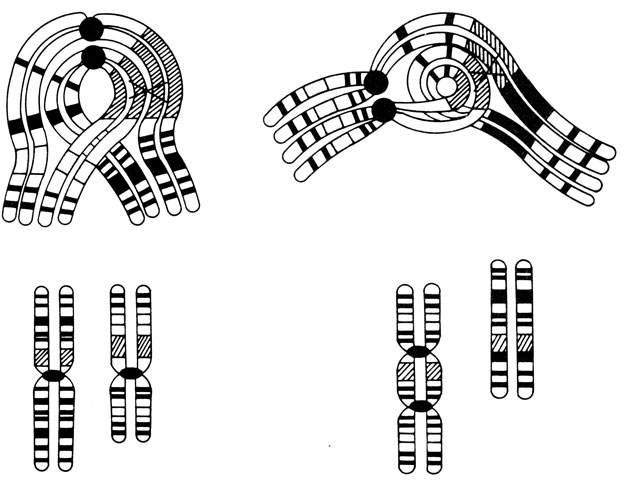
Рис. 2.39. Нарушение спаривания хромосом во время мейоза у гетерозиготы по перицентрической (слева) и парацентрической (справа) инверсии. Предполагают, что в обоих случаях кроссинговер происходит в сегментах, отмеченных на рисунке крестом. Вследствие разрывов и обменов образуются аномальные хромосомы, что в свою очередь приводит к анеуплоидии зиготы в ближайшем поколении
Другой тип внутренних обменов представляют кольцевые хромосомы (рис. 2.40). Перестройка этого типа возникает при утрате обоих теломерных участков хромосомы (как ацентрических фрагментов) и последующем воссоединении открытых концов. Судьба кольцевой хромосомы в митозе зависит от того, как завершилось воссоединение концов сестринских хроматид. Если во время репликации ДНК обмен между сестринскими нитями в точках разрыва не происходит, то кольцо, удваиваясь, образует два отдельных кольца, каждое со своей центромерой. Такие кольцевые хромосомы проходят через митоз без затруднений. Один обмен между сестринскими нитями ведет к образованию большого кольца с двумя центромерами. Дицентрическая структура обычно разрушается в наступающем митозе. Два обмена могут привести к образованию двух колец, "сцепленных" друг с другом подобно звеньям цепи. Детали различных вариантов представлены на рис. 2.40. Иногда хроматидные разрывы и образование колец происходят в фазе G2, и тогда в отдельной клетке наблюдается картина, показанная на рис. 2.41.
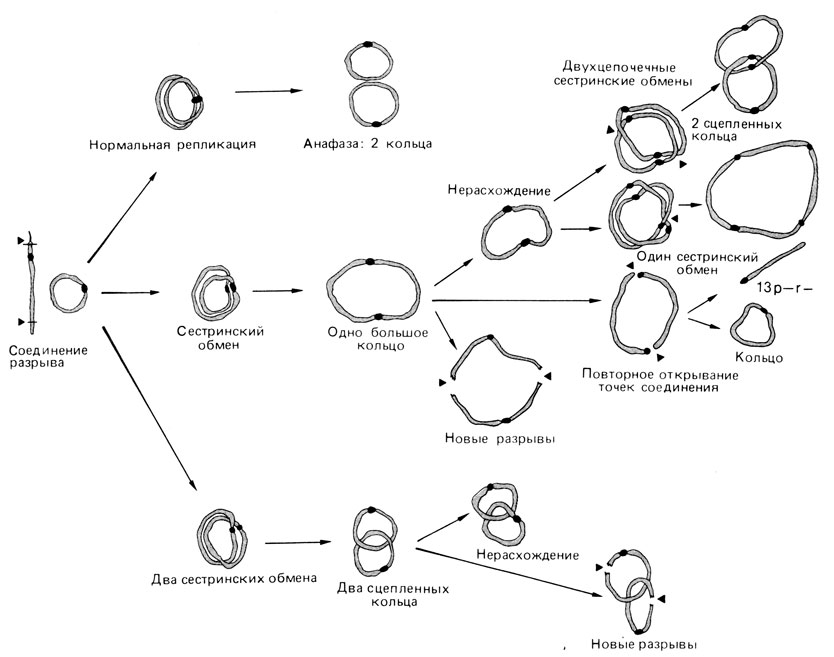
Рис. 2.40. Образование кольцевой хромосомы в фазе G1 и судьба такой хромосомы в митозе (r- - означает отсутствие кольца)
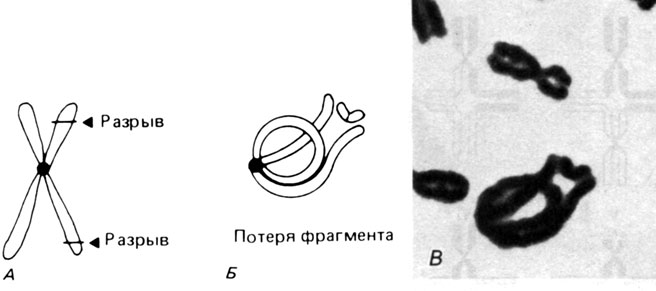
Рис. 2.41. Образование кольцевой хромосомы в фазе G2. А. Два разрыва в одной из двух сестринских хроматид. Б. Воссоединение разорванных концов; спаривание фрагментов с гомологичными хроматидными сегментами. В. Та же кольцевая хромосома в метафазе человека. (Courtesy of, Dr.T.M. Schroeder-Kurth.)
Межхромосомные перестройки (внешние обмены). Во многих случаях воссоединение открытых концов затрагивает разные хромосомы как гомологичные, так и негомологичные. Если разрыв происходит в фазе Gl, то воссоединение обычно завершается в той же фазе G1 (или ранней S) перед репликацией ДНК. Если каждая из перестроенных хромосом сохраняет центромеру, то такие транслокационные хромосомы могут пройти через наступающий митоз без всяких затруднений. Если одна из перестроенных хромосом приобретает две центромеры, формируется дицентрическая хромосома. В зависимости от деталей репликации она может пройти через наступающий митоз при следующих условиях: 1) если обе центромеры отойдут к одному и тому же полюсу и 2) если репликация и сестринский хроматидный обмен между двумя центромерами не приведут к переплетению хроматид (рис. 2.42). Если разрывы и воссоединения концов завершатся после репликации ДНК, то затронутой окажется только одна сестринская хроматида каждой хромосомы. Воссоединенные сестринские хроматиды еще остаются спаренными с их неповрежденными партнерами. Это ведет к межхромосомным обменам, которые обнаруживаются в первом митотическом делении после воссоединения. Различные типы этих обменов показаны на рис. 2.43.
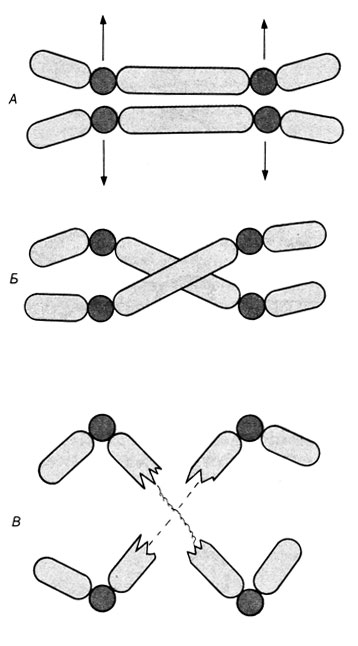
Рис. 2.42. Дицентрическая хромосома в анафазе митоза. А. Обе центромеры направляются к одному и тому же полюсу; хромосомы остаются интактными. Б. Центромеры направляются к противоположным полюсам. Образуются анафазные мосты. В. Хромосомы разрываются
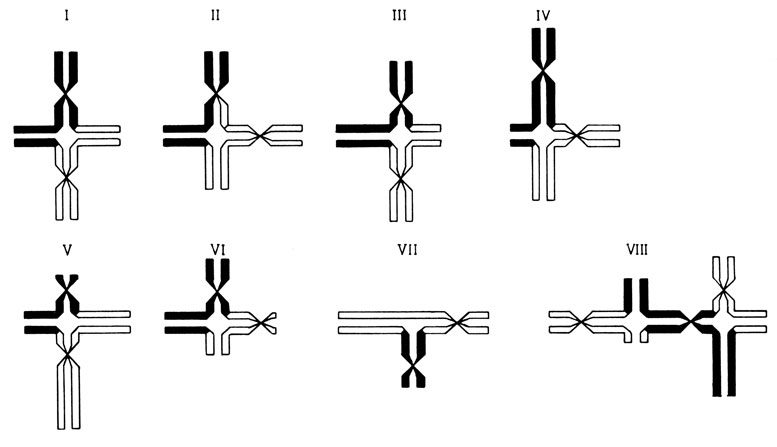
Рис. 2.43. Классы обменов, возникающих после транс локации в фазе G2. В обмене участвуют две гомологичные хромосомы. I. Альтернативное положение центромер; обмен фрагментами равной длины. II. Смежное положение центромер; обмен фрагментами равной длины. III. Альтернативное положение центромер; обмен фрагментами разной длины. IV. Смежное положение центромер; обмен неравными фрагментами. В обмене участвуют две негомологичные хромосомы. V. Альтернативное положение центромер. VI. Смежное положение центромер. VII. Трирадиальная конфигурация (необходима утрата фрагментов). Комплекс негомологичных хромосом (больше двух). VIII. Пример фигур с тремя хромосомами
Если каждая из перестроенных хромосом сохранит центромеру (рис. 2.43; класс I, III и V), то анафазное расхождение хроматид в обеих таких хромосомах будет протекать без всяких затруднений. Однако если обе центромеры окажутся в одном и том же сегменте, то образующиеся дочерние клетки в любом случае будут анеуплоидными: либо центромеры отойдут к разным полюсам и возникнет "анафазный мост", который приведет в конце концов к разрыву, либо две центромеры отойдут к одному и тому же полюсу. В этом случае перестройка завершится только негомологичным воссоединением (рис. 2.43, классы VI, VII). Дальнейшие события откладываются до следующего митоза, в котором появляется дицентрическая хромосома. Иногда она может пройти и этот митоз. В любом случае, однако, при указанных выше условиях межхромосомные обмены, как правило, приводят к гибели клеток вследствие анеуплоидии или нарушений в митозе.
В соматических тканях человека многие из этих митотических нарушений можно видеть даже в рутинных клеточных препаратах, приготовленных без использования специальных хромосомных методик. На рис. 2.44, А и Б показаны анафазные мосты и так называемые микроядра в клетках костного мозга человека. Микроядра формируются теми хромосомами (или хромосомными фрагментами), которые не связаны с митотическим аппаратом и не принимают участия в митозе, как остальные хромосомы. Это сопровождается преждевременной конденсацией таких хромосом и их фрагментов. В метафазных хромосомах основного ядра хроматиды имеют обычно нормальную степень конденсации, в то время как хромосомы микроядра конденсированы по типу профазных (рис. 2.45). Эти цитологические феномены полезно использовать для экспресс-оценки мутагенных агентов (разд. 5.2). Преждевременную конденсацию хромосом можно увидеть также in vitro при слиянии интерфазной клетки с другой, находящейся в предмитотической фазе [500]. Этот метод пригоден для изучения строения хромосом в интерфазном ядре.
![Рис. 2.44. А. Образование микроядер вследствие хромосомных аберраций в костном мозге больного с анемией Фанкони. Б. Анафазный мост, образованный дицентрической хромосомой (у того же больного) [212]](pic/000052.jpg)
Рис. 2.44. А. Образование микроядер вследствие хромосомных аберраций в костном мозге больного с анемией Фанкони. Б. Анафазный мост, образованный дицентрической хромосомой (у того же больного) [212]
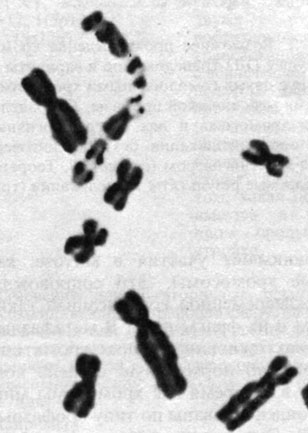
Рис. 2.45. Преждевременная конденсация хромосом: профазоподобные хромосомы из микроядер вместе с некоторыми другими, нормальными метафазными хромосомами. (Courtesy of Dr.T.M. Schroeder-Kurth.)
Транслокации могут привести к нарушениям и в мейозе, так как на ранних стадиях этого деления гомологичные хромосомы конъюгируют. Если в перестройке участвуют три хромосомы, как, например, у носителей сбалансированных транслокаций D/G или G/G, в метафазе I они образуют так называемые трехзвенные цепочки. На рис. 2.46 показана такая структура у носителя сбалансированной D/D-транслокации. На рис. 2.46, А и Б представлена схема событий, которые происходят с этой хромосомой на стадии пахитены в процессе кроссинговера; на рис. 2.46, В - картина, которую можно ожидать в диакинезе, если каждая из двух свободных хромосом имеет один перекрест с транслокационной хромосомой. Для сравнения показана трехзвенная цепочка в том виде, как она наблюдается реально. Если в перестройку вовлечены четыре хромосомы, то образуется четырехзвенная цепочка. Такое событие ведет иногда к последующей анеуплоидии в зависимости от комбинаторики анафазных движений четырех центромер. Если две центромеры одного элемента отходят к одному полюсу и если хроматиды не переплетутся между центромерами, то деление завершится нормальной анафазой. Однако очень часто происходят дополнительные разрывы хромосом. Мейоз служит хорошим фильтром для хромосомных перестроек.
![Рис. 2.46. А. Возможное происхождение хромосомы, несущей D/D-транслокацию и варианты ее спаривания с двумя гомологичными хромосомами во время мейотической профазы. Б. Транслокационная хромосома и два ее гомологичных партнера после репликации без кроссинговера (слева) и с кроссинговером (справа). В. Теоретически ожидаемые результаты транслокации (тривалент) в диакинезе и метафазе I (слева) и сравнение их с действительно наблюдаемой картиной (справа). Г. Хромосомы, участвующие в образовании тривалента, вероятно, распределяются по дочерним клеткам на два класса, дающие разные типы сбалансированных (I) и несбалансированных (II) гамет [405]](pic/000054.jpg)
Рис. 2.46. А. Возможное происхождение хромосомы, несущей D/D-транслокацию и варианты ее спаривания с двумя гомологичными хромосомами во время мейотической профазы. Б. Транслокационная хромосома и два ее гомологичных партнера после репликации без кроссинговера (слева) и с кроссинговером (справа). В. Теоретически ожидаемые результаты транслокации (тривалент) в диакинезе и метафазе I (слева) и сравнение их с действительно наблюдаемой картиной (справа). Г. Хромосомы, участвующие в образовании тривалента, вероятно, распределяются по дочерним клеткам на два класса, дающие разные типы сбалансированных (I) и несбалансированных (II) гамет [405]
Хромосомные разрывы имеют место как в соматических, так и в половых клетках. Изучение этих явлений в соматических клетках актуально с точки зрения мутационных исследований (разд. 5.2). Разрывы хромосом в половых клетках могут передаваться следующему поколению, что часто приводит к гибели зиготы на эмбриональной стадии. Однако в некоторых случаях хромосомная аберрация оказывается совместимой с постнатальной жизнью, и это приводит к рождению ребенка с хромосомным синдромом. Прежде чем перейти к анализу некоторых из этих синдромов, необходимо описать общепринятую номенклатуру кариотипа человека. Эта номенклатура была разработана группой цитогенетиков и согласована на Парижской конференции в 1971 г. [468].
Описание кариотипа человека. При описании кариотипа человека прежде всего указываются общее число хромосом и набор половых хромосом. Затем отмечается, какая хромосома лишняя, какой не хватает, а также структурно измененные. Некоторые примеры представлены в табл. 2.4.

Таблица 2.4
![Таблица 2.5. Номенклатурные символы, дополнительные к тем, которые рекомендованы Чикагской конференцией (1966). (Парижская конференция, 1971 [468].)](pic/000057.jpg)
Таблица 2.5. Номенклатурные символы, дополнительные к тем, которые рекомендованы Чикагской конференцией (1966). (Парижская конференция, 1971 [468].)
Делеционные синдромы. Индивид, гетерозиготный по делеции, является моносомиком по соответствующему району хромосомы. Де Груши и сотр. (1963) [367] первыми описали делецию del 18p-, однако делеционный синдром впервые был обнаружен Леженом и сотр. в 1963 г. [418]. Ими были выявлены трое детей с делецией короткого плеча хромосомы 5 (del 5p-). Кроме обычных признаков аутосомных аномалий (общее отставание в развитии и низкий вес при рождении) у этих детей отмечалось лунообразное лицо с гипертелоризмом (широко расставленные глаза). Во внешнем облике больных не было каких-то ярких особенностей (рис. 2.47), однако их плач напоминал мяуканье кошки (cri du chat или cat cry).
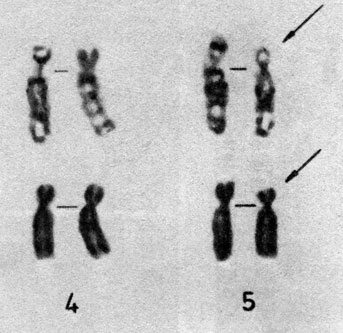
Рис. 2.47. Часть кариотипа в случае синдрома кошачьего крика - делеция 5p
Существует несколько разных механизмов возникновения делеций и соответственно разные типы самих делеций: 1) истинная концевая делеция, 2) интерстициальная делеция и 3) делеция в результате транслокации. Во многих сообщениях указывается на наличие при синдроме "кошачьего крика" транслокации. На рис. 2.48 представлена часть кариотипа пробанда с хромосомой 5p-. Делетированный участок включает 5p15 и часть 5p16 сегментов. У фенотипически нормальной матери обнаружена такая же хромосома 5, но одна из хромосом 17-й пары имела лишний сегмент на длинном плече между 17ql2 и 17q21. Следовательно, концевой сегмент хромосомы 5 содержится в длинном плече хромосомы 17. Случай, выявленный при помощи G-метода (рис. 2.47), иллюстрирует пример истинной концевой делеции.
![Рис. 2.48. А. Часть кариотипа больного с синдромом кошачьего крика, имеющего делению концевого сегмента хромосомы 5. Б. Сбалансированная транслокация у матери этого больного. Концевой сегмент хромосомы 5 встроен в хромосому 17 [300]](pic/000058.jpg)
Рис. 2.48. А. Часть кариотипа больного с синдромом кошачьего крика, имеющего делению концевого сегмента хромосомы 5. Б. Сбалансированная транслокация у матери этого больного. Концевой сегмент хромосомы 5 встроен в хромосому 17 [300]
Внутренние обмены: парацентрические и перицентрические инверсии [401а]. Парацентрические инверсии (т. е. не вовлекающие центромеру) у человека обнаруживаются с большим трудом. Они будут обсуждаться в контексте хромосомной эволюции (разд. 7.2.1). Начиная с 60-х гг. было опубликовано много работ о предполагаемых перицентрических (т. е. захватывающих центромеру) инверсиях. У некоторых носителей таких инверсий выявлены различные аномалии типа умственной отсталости или пороков развития. Фенотип других не обнаруживал каких-либо заметных отклонений, но в браках с ними регистрировались повторные спонтанные аборты. У представителей третьей группы не обнаружено вообще никаких аномалий. Следует отметить, что при использовании обычных методов окрашивания хромосом перицентрические инверсии выявляются относительно редко.
С внедрением в широкую практику методов дифференциального окрашивания появились сообщения о более высокой частоте инверсий в некоторых популяциях. Довольно часто в эти перестройки вовлекается хромосома 9. Именно такая ситуация обнаружена в Финляндии [323]. При анализе кариотипов 631 жителя этой страны по различным диагностическим поводам у 9 была обнаружена перицентрическая инверсия и в 6 случаях - в хромосоме 9. Все эти 6 инверсий оказались идентичными. У трех пробандов инверсия была обнаружена в хромосоме 10: при этом у двух одинаковая, а у третьего отличная от них. Инверсию в хромосоме 9 может легко распознать и неспециалист (рис. 2.49), так как типичная вторичная перетяжка оказывается в этом случае не в длинном, а в коротком плече. Идентификация инверсии в хромосоме 10 требует особого опыта (рис. 2.50).
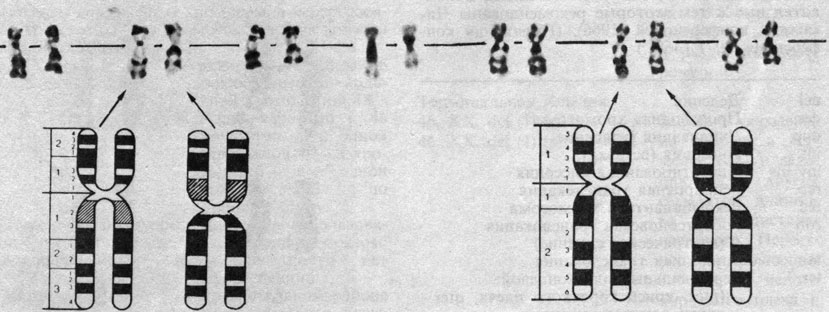
Рис. 2.49. Хромосома 9 от разных гетерозигот по одинаковой инверсии inv 9 (pl; q13). В каждой паре слева помещен нормальный гомолог, справа - инвертированная хромосома, три препарата окрашены по G-методу, один - по C-методу. Две правые пары хромосом принадлежат индивиду со вторичной перетяжкой в нормальном гомологе. Рис. 2.50. Хромосома 10 от разных индивидов, гетерозиготных по одинаковой инверсии inv (pl; q21). В каждой паре нормальный гомолог помещен слева, а инвертированная хромосома - справа. Препараты окрашены G-методом
При исследовании мейоза у двух пробандов с инверсией хромосомы 9 идентифицированный бивалент 9 имел нормальную морфологию, но около вторичной перетяжки не было выявлено ни одной хиазмы. Весьма вероятно, что инверсии приводят к несовершенной конъюгации и к подавлению кроссинговера, как это хорошо известно на примере других организмов, в частности у дрозофилы. Подобные инверсии можно использовать для региональной локализации генов соответствующих районов хромосомы 9 (разд. 3.4). Эти инверсии не влияют на мейотическую сегрегацию хромосом и не приводят к пренатальной гибели гетерозигот, как это следует из специальных работ (см. разд. 3.3): в браках между нормальной гомозиготой и гетерозиготой по inv (9) 25 потомков имели нормальный кариотип, 23 - были гетерозиготами. Аналогично для двух типов inv (10) суммарно это отношение оказалось равным 10:11. Еще в одном браке между двумя гетерозиготами по инверсии, оказавшимися дальними родственниками, среди детей обнаружена одна гомозигота. Такого рода наблюдения проливают свет на механизмы хромосомной эволюции (разд. 7.2.1).
Как отмечалось выше, пробанды в этом исследовании направлялись на консультацию с диагностическими целями, поэтому вряд ли неожиданным является тот факт, что у них выявлены разнообразные аномалии. Однако эти аномалии трудно было охарактеризовать как единый синдром. Более того, среди родственников с инверсиями были и вполне нормальные в клиническом отношении. Следовательно, весьма вероятно, что инверсии в хромосомах 9 и 10 не влияют ни на фенотип носителей, ни на их плодовитость.
Перицентрические инверсии обнаружены и в хромосоме 2 (рис. 2.51) [419]. В этом сообщении речь идет о трех семьях. Две из них обследованы по поводу рождения детей с пороками развития, тогда как третья - по поводу привычных выкидышей, т. е. эта выборка является сильно смещенной и возможность того, что привычные выкидыши могут быть вызваны инверсиями, исключить нельзя. Исследуя происхождение семей этих пробандов в разных странах, авторы указывают, что вряд ли данная инверсия имеет общее происхождение. Они ссылаются на случай, где та же инверсия описана как новая мутация [383]. Более вероятным им представляется предположение о повышенной ломкости хромосомы в соответствующем сегменте. Однако ссылка на новую мутацию была привлечена до того, как стали применяться методы дифференциального окрашивания.
![Рис. 2.51. Перицентрическая инверсия в хромосоме 2. А. G-окрашивание. Б. Схематическое изображение сегментации [419]](pic/000060.jpg)
Рис. 2.51. Перицентрическая инверсия в хромосоме 2. А. G-окрашивание. Б. Схематическое изображение сегментации [419]
Очень маленькие инверсии могут встречаться в отдельных популяциях довольно часто, так как скорее всего они совершенно не влияют на состояние здоровья или плодовитость. Если инверсия затрагивает протяженный участок хромосомы, то возможность нарушений в мейозе более вероятна. Однако сами носители инверсий эуплоидны, поэтому вряд ли следует ожидать у них какие-либо фенотипические аномалии.
Рекомбинационная анеусомия. Встречаются семьи, в которых один из родителей, по-видимому, имеет такую же аберрацию, что и ребенок, например перицентрическую инверсию или транслокацию. При этом родитель фенотипически нормален, в то время как у ребенка обнаруживается тяжелый синдром нарушения развития. Факты такого рода можно объяснить случайным сочетанием в одной семье наследуемого полиморфного хромосомного варианта и каких-то нарушений развития различной этиологии. Однако в других случаях кроссинговер в участке инверсии или транслокации между аномальной хромосомой и ее нормальным гомологом может вести к появлению несбалансированных наборов хромосом в половых клетках. Такое объяснение было выдвинуто Леженом и Берже еще в 1965 г. [416], но его подтверждение получили только после появления методов дифференциального окрашивания.
Впервые указанный механизм удалось продемонстрировать реально в работе [340]. Речь идет о мальчике с множественными пороками развития. На рис. 2.52 показаны хромосомы 10 этого пробанда и его матери. Можно видеть, что у матери имеется большая перицентрическая инверсия. Кроссинговер в пределах этой инверсии привел к появлению аномальной хромосомы, в результате чего ребенок оказался трисомиком по сегменту q456. Без применения метода дифференциального окрашивания все С-хромосомы (группа 6-X-12) были бы классифицированы как нормальные и кариотипы матери и ребенка рассматривались бы как идентичные. Высокоразрешающие методы позволяют выявлять такие случаи.
![Рис. 2.52. Рекомбинационная анеусомия. Кариотип пробанда с пороками развития и его нормальной матери (Q- и G-окрашивание). Пояснение в тексте. А. Часть кариотипа с хромосомой 10 матери. Б. Часть кариотипа с хромосомой 10 сына [340]](pic/000061.jpg)
Рис. 2.52. Рекомбинационная анеусомия. Кариотип пробанда с пороками развития и его нормальной матери (Q- и G-окрашивание). Пояснение в тексте. А. Часть кариотипа с хромосомой 10 матери. Б. Часть кариотипа с хромосомой 10 сына [340]
Кольцевые хромосомы. Иная ситуация характерна для кольцевых хромосом. Поскольку образование кольца, как полагают, связано с утратой теломерных сегментов хромосомы, носители кольцевых хромосом должны напоминать носителей соответствующих делеций. Например, если в кольцевую перестройку вовлечена хромосома 5p, у пробанда может наблюдаться синдром "кошачьего крика" [455]. В других случаях в зависимости от размеров делегированного участка симптомы могут быть менее выраженными.
Так, например, кольцевая хромосома 13 была обнаружена у 14-месячного ребенка с умственной отсталостью и такими признаками, как микроцефалия, эпикант, широкая спинка носа, выступающие ушные раковины, микрогнатия [382]. В 85% лимфоцитов крови и в 82% фибробластов кожи выявлялось простое кольцо, идентифицированное как 13r (pll; q34). В 7% лимфоцитов и в 6% фибробластов можно было наблюдать двойное дицентрическое кольцо, которое состояло из двух хромосом 13. В 5% лимфоцитов и в 8% фибробластов кольцо отсутствовало, одна метафаза была с двумя сцепленными двойными кольцами, остальные клетки содержали другие аномалии. На рис. 2.40 показана судьба кольцевой хромосомы в митозе. В большинстве случаев кольцо реплицируется и проходит через митоз нормально. Иногда происходит один сестринский обмен и формируется двойное кольцо с двумя центромерами. Двойной сестринский обмен может привести к образованию двух сцепленных колец. В следующей интерфазе двойное кольцо может снова претерпеть один, два или более сестринских обмена, что в свою очередь приведет к двойным сцепленным кольцам или к четверным кольцам. Таким образом, возможно большое число разных вариантов. На рис. 2.53 представлено двойное сцепленное кольцо, на рис. 2.54, А - четверное кольцо. Многие из этих заново формирующихся колец ведут к нарушениям в митозе вследствие все большего числа разрывов и последующей анеуплоидии в дочерних клетках. На рис. 2.55 показана анафаза с разрывами дицентрических колец на равные и неравные части. Большая часть теоретически возможных конфигураций (рис. 2.40) действительно наблюдалась в данном случае.
![Рис. 2.53. Рисунок сегментации моноцентрической (слева) и дицентрической (справа) кольцевой хромосомы [382]](pic/000062.jpg)
Рис. 2.53. Рисунок сегментации моноцентрической (слева) и дицентрической (справа) кольцевой хромосомы [382]
![Рис. 2.54. А. Два соединенных двойных кольца из диплоидной клетки рядом с другой хромосомой 13-й пары. Б. Тетрацентрическое кольцо из тетраплоидной клетки [382]](pic/000063.jpg)
Рис. 2.54. А. Два соединенных двойных кольца из диплоидной клетки рядом с другой хромосомой 13-й пары. Б. Тетрацентрическое кольцо из тетраплоидной клетки [382]
![Рис. 2.55. Дицентрическое кольцо может разорваться на две равные (Б, Г) или две неравные (А, В) части [382]](pic/000064.jpg)
Рис. 2.55. Дицентрическое кольцо может разорваться на две равные (Б, Г) или две неравные (А, В) части [382]
Фрагменты. Хромосомные фрагменты, не содержащие центромеры или ее части (так называемые ацентрические фрагменты), в митозе и мейозе обычно теряются, но при наличии центромеры они могут сегрегировать как дополнительные, маркерные, хромосомы. При исследовании случайной выборки новорожденных в Дании (разд. 5.1.2.1) такие маркеры оказались не редкими; в некоторых случаях у носителей этих маркерных хромосом обнаруживаются фенотипические аномалии.
Изохромосомы. Иногда выявляются хромосомы, оба плеча которых идентичны. Их называют изохромосомами. Можно предположить, что они возникают вследствие аномального разделения метафазной хромосомы, как показано на рис. 2.56. Если в такую перестройку вовлекается неравноплечая хромосома, то может образоваться изохромосома и по короткому, и по длинному плечу. Относительно часто наблюдаются изохромосомы X. В случае изохромосомы по длинному плечу X, i (Xq) развивается синдром Тернера, поскольку данная хромосома всегда инактивирована и активной остается только одна нормальная X-хромосома (разд. 2.2.3).
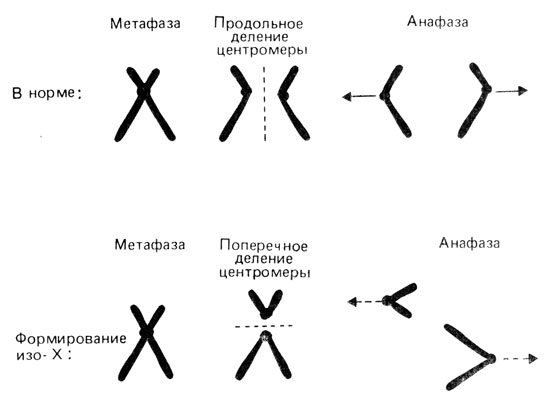
Рис. 2.56. Образование изохромосом путем разделения центромеры
Межхромосомные обмены: центрические слияния (робертсоновские транслокации). Центрическое слияние является наиболее частым типом хромосомных перестроек в человеческих популяциях. Первые описанные случаи транслокационного синдрома Дауна были связаны с центрическим слиянием между длинным плечом хромосомы 21 и одной из D- или G-хромосом. Впоследствии о таких больных сообщалось неоднократно. Среди всех случаев синдрома Дауна транслокации этого типа составляют всего лишь несколько процентов, и многие из них являются вновь возникшими. Важно, что в центрическое слияние могут вовлекаться все пять пар акроцентрических хромосом. Короткие плечи этих хромосом содержат ядрышковые организаторы, в частности гены рРНК (разд. 2.3). При этом в интерфазном ядре короткие плечи, включая центромерные районы, располагаются в тесной близости от ядрышка. Благодаря применению методов дифференциального окрашивания появилась возможность исследовать участие отдельных D- и G-хромосом в центрических слияниях. Оказалось, что оно не является случайным (табл. 2.6). Данные, представленные в этой таблице, основаны на исследовании новорожденных. Следует учесть, что результаты могут быть искажены из-за неодинаковой частоты эмбриональной смертности в различных группах. Центрическое слияние означает, что короткие плечи двух акроцентрических хромосом и, вероятно, одна из центромер утрачены (рис. 2.57), т. е. утрачены также и гены рибосомной РНК. Действительно, по данным ДНК - РНК-гибридизации среднее число генов рРНК меньше у так называемых сбалансированных носителей центрических слияний, чем в общей популяции [1020, 1061]. Однако это не приводит к каким-либо функциональным различиям, и носители таких хромосом совершенно здоровы.
![Таблица 2.6. Хромосомы, вовлеченные в робертсоновские транслокации. (Семейный материал, проанализированный Шефером [501а].)](pic/000066.jpg)
Таблица 2.6. Хромосомы, вовлеченные в робертсоновские транслокации. (Семейный материал, проанализированный Шефером [501а].)
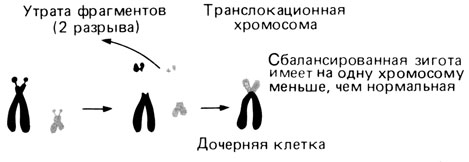
Рис. 2.57. Принцип центрического слияния (робертсоновская транслокация). Две акроцентрические хромосомы утратили свои короткие плечи, а длинные плечи слились. Транслокационная хромосома может иметь одну или две центромеры; в последнем случае одна из центромер может быть супрессирована. В любом случае у сбалансированной гетерозиготы количество хромосом будет на единицу меньше, чем у нормального индивида. (При реципрокной транслокации образуется сбалансированная зигота с нормальным числом хромосом.)
На рис. 2.58 показаны возможные комбинации хромосом в половых клетках носителя D/G- и G21/G21-транслокации. После оплодотворения нормальным сперматозоидом возможны шесть разных вариантов. Однако первые два - моносомия D и трисомия D- никогда не наблюдались, а моносомия 21 по крайней мере в большей части известных случаев летальна. Каждый из остальных трех вариантов - трисомия 21, сбалансированная транслокация и нормальный набор - ожидается с вероятностью 1/3. Это ожидание, однако, не подтверждается на практике: когда мать является носителем, вероятность составляет около 15%, а если носитель отец, вероятность не превышает 5%. Однако риск появления сбалансированной транслокации составляет, как и ожидается, ∼50%.
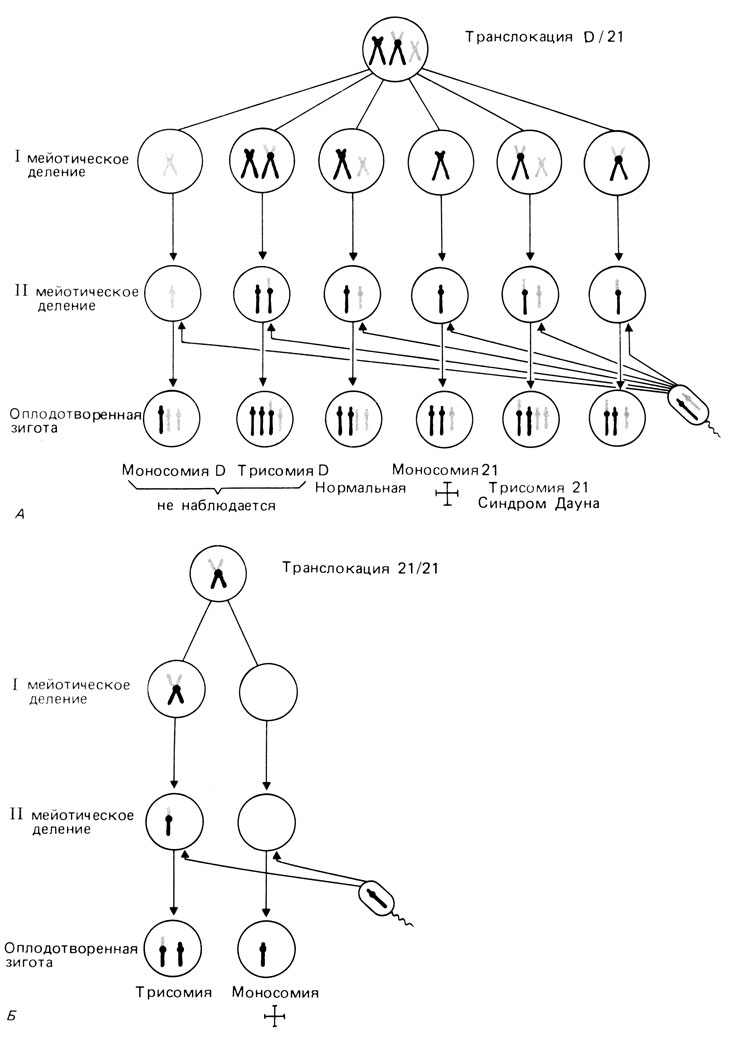
Рис. 2.58. А. Схема образования половых клеток у женщины - носительницы сбалансированной транслокации D/21: одна D-хромосома приобретает транслоцированное длинное плечо хромосомы 21. В результате остается только одна свободная хромосома 21. Поскольку эта свободная хромосома 21 и две D-хромосомы комбинируются случайно, теоретически может образоваться шесть разных типов гамет и после оплодотворения нормальным сперматозоидом соответственно шесть разных типов зигот. Однако три типа из шести возможных не обнаруживаются. Остальные индивиды либо нормальны, либо имеют сбалансированный кариотип, либо трисомики. Б. Образование половых клеток с транслокацией 21/21 и 21-изохромосомой. Существует две возможности: если транслоцированная хромосома попадет в половую клетку, то зигота окажется функционально трисомной и у ребенка будет синдром Дауна; в том случае, если транслокационная хромосома не попадет в половую клетку, зигота будет лишена хромосомы 21 и погибнет
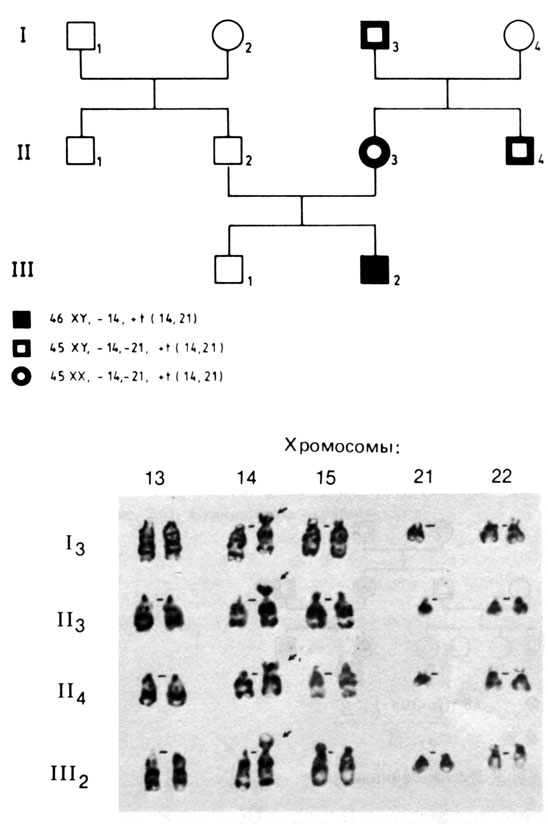
Рис. 2.59. Родословная ребенка с синдромом Дауна и робертсоновской транслокацией 14/21 (III, 2). Мать (II, 3), ее брат (II, 4) и дед по материнской линии (I, 3) имеют сбалансированный кариотип. (Courtesy of Dr.T.M. Schroeder-Kurth.)
При транслокации 21/21 (как и в случае 21/21 изохромосомы) прогнозы намного более мрачные: либо ребенок будет трисомиком с синдромом Дауна, либо анеуплоидия будет летальной. К счастью, в настоящее время можно при помощи методов дифференциального окрашивания отличить транслокацию 21/21 от транслокации 21/22, при которой вероятность анеуплоидных зигот намного меньше - такая же, как и в случае D/G-транслокаций.
Межхромосомные обмены: реципрокные транслокации. В отличие от центрических слияний реципрокные транслокации не обязательно связаны с утратой материала. Фрагменты хромосом воссоединяются в новых комбинациях, но с сохранением в зиготе эуплоидного числа 46, а не 45, как при центрических слияниях. На рис. 2.60 представлены типы дочерних клеток, которые можно ожидать в случае реципрокных транслокаций. Чаще всего выявляются только частичные трисомики и частичные моносомики. Другие комбинации, как полагают, лета льны.
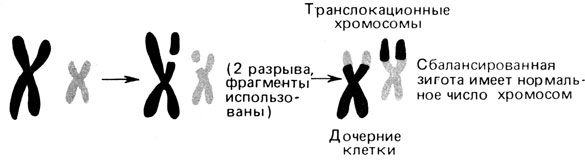
Рис. 2.60. Реципрокная транслокация. Сбалансированная зигота имеет 46 хромосом; в двух хромосомах видны комплементарные структурные аномалии
Типичный случай описан в работе [504]. На рис. 2.61 показаны два умственно отсталых сибса в возрасте 11 и 9 лет. В их фенотипе обнаружены как конкордантные, так и дискордантные признаки (табл. 2.7). При исследовании кариотипа обычным методом у обоих детей выявлено удлинение длинного плеча одной из С-хромосом (рис. 2.62); у матери и бабки (по линии матери) обнаружена такая же хромосома и, кроме того, другая аномальная хромосома в группе С (6-X-12), у которой почти полностью отсутствовало короткое плечо (рис. 2.63). С помощью G-метода у матери выявлена реципрокная транслокация между хромосомами 7 и 10, кариотип 46, XX, t (7; 10) (p22; pll). Результатом такой перестройки является частичная трисомия 10p + у обоих детей. Особенность данного случая заключается не только в конкордантности многих признаков у обоих детей, что укладывается в единый клинический синдром, но и в наличии ряда дискордантных симптомов, что указывает на изменчивость фенотипических аномалий, вызванных одной и той же хромосомной аберрацией.
![Таблица 2.7. Конкордантные и дискордантные признаки [504]](pic/000069.jpg)
Таблица 2.7. Конкордантные и дискордантные признаки [504]
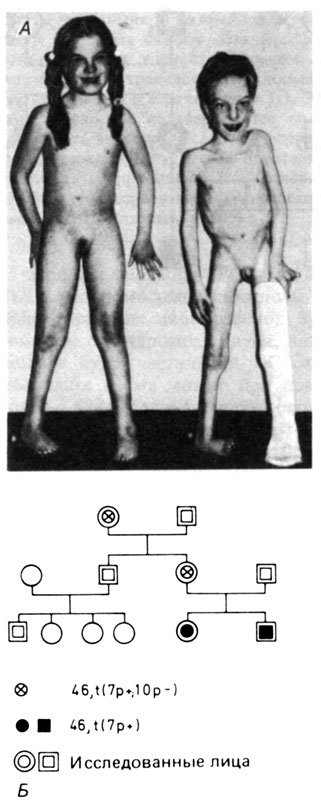
Рис. 2.61. А. Брат и сестра с тяжелыми пороками развития и умственной отсталостью. Б. Родословная этих сибсов (мать несет сбалансированную транслокацию; дети - частичные трисомики 10p +)
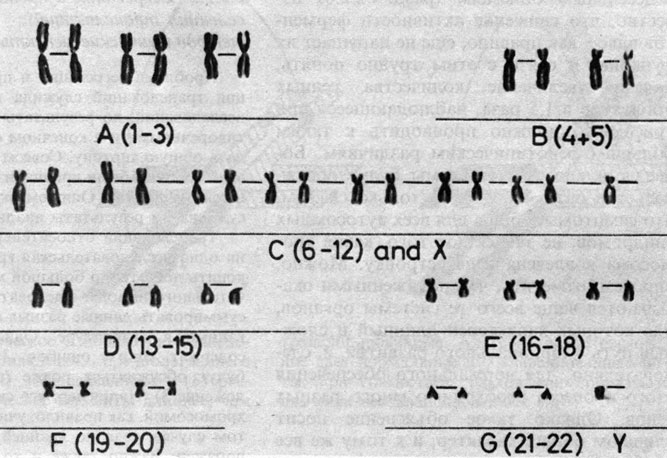
Рис. 2.62. Кариотип мальчика, показанного на рис. 2.61. Стандартное окрашивание
![Рис. 2.63. Кариотип матери двух детей, показанных на рис. 2.61, с реципрокной транслокацией, в которой участвуют хромосомы 7 и 10. Эти две хромосомы указаны стрелками (G-окрашивание) [504]](pic/000074.jpg)
Рис. 2.63. Кариотип матери двух детей, показанных на рис. 2.61, с реципрокной транслокацией, в которой участвуют хромосомы 7 и 10. Эти две хромосомы указаны стрелками (G-окрашивание) [504]
Основные фенотипические проявления аутосомных аберраций. Наиболее заметной особенностью фенотипов при аутосомных аберрациях является очень частое совпадение многих признаков и симптомов.
Основные признаки: а) Общие низкий вес при рождении резкая задержка развития умственная отсталость (обычно тяжелая) низкий рост б) Голова и лицо микроцефалия неполная оссификация микрогнатия аномальное расположение глаз "дизморфическое лицо" низко расположенные и деформированные ушные раковины в) Верхние и нижние конечности аномальный дерматоглифический рисунок г) Внутренние органы врожденный порок сердца и/или крупных сосудов пороки развития мозга пороки развития мочеполовой системы
Следующие признаки обычно не указываются как характерные для аутосомных аномалий и описываются как исключения:
умственная отсталость без каких-либо пороков развития пороки развития при нормальном психическом развитии изолированные (одиночные) пороки развития.
При многих, хотя и не при всех аутосомных аберрациях, кроме этих общих пороков развития находят более или менее специфичные. Важно отметить, что и общие признаки могут проявляться с большей или меньшей тяжестью. Ряд признаков, причиной которых является специфическая аберрация, обычно формируют неслучайное сочетание, характерное именно для данной аберрации. Подозрение на хромосомный дефект может возникнуть при клиническом обследовании, но ставится диагноз только на основе хромосомного анализа. Наличие характерных симптомов указывает на необходимость исследования хромосом.
У разных больных с одинаковой аберрацией многие характеристики одного синдрома сильно варьируют. Так, при синдроме Дауна, например, в ряде случаев умственная отсталость может быть выражена в незначительной степени, в то время как в большинстве случаев наблюдается олигофрения тяжелой степени; кроме того, пороки сердца находят у многих таких больных, а атрезию кишечника - крайне редко. У сибсов на рис. 2.61 с одной и той же транслокацией кроме некоторых общих черт обнаруживаются и явные фенотипические различия. Можно предположить, что эта изменчивость зависит частично от того, что у разных индивидов одна и та же аномальная хромосома проявляет свои эффекты на разном генетическом фоне.
Наиболее неожиданный факт, касающийся фенотипа хромосомных аберраций, состоит в том, что при трисомиях вообще обнаруживаются аномалии. Ведь носители этих аберраций имеют полный набор генетического материала, и ни один из генов не утерян и не поврежден! По данным исследований гетерозигот по аутосомным рецессивным болезням (разд. 4.2.2.8) известно, что снижение активности ферментов вдвое, как правило, еще не нарушает их функции. В связи с этим трудно понять, почему увеличение количества генных продуктов в 1,5 раза, наблюдающееся при трисомиях, должно приводить к таким большим фенотипическим различиям. Более детально эти проблемы будут обсуждаться в разд. 4.7.4. Здесь только скажем, что симптомы, общие для всех аутосомных синдромов, не зависят от того, какая хромосома вовлечена в перестройку. Можно, впрочем, отметить, что пораженными оказываются чаще всего те системы органов, для которых характерен длинный и сложный путь эмбрионального развития, и, следовательно, для нормального обеспечения этого процесса необходимо много разных генов. Однако такое объяснение носит слишком общий характер, и к тому же все необходимые гены имеются в наличии.
Чем же можно объяснить нарушения, вызываемые хромосомными аберрациями? Ответ таков: эти синдромы обусловлены, вероятно, не наличием избыточной активности или дефекта отдельных генов, а главным образом нарушениями регуляции активности генов во время эмбрионального развития. Следовательно, анализ аутосомных аберраций может оказаться полезным для понимания механизмов генной регуляции у человека. В случае такой специальной проблемы, как развитие половых признаков, изучение больных с численными и структурными аберрациями половых хромосом оказалось весьма поучительным. Однако до подробного обсуждения аберраций этого типа полезно сделать несколько замечаний относительно сегрегации и пренатальной селекции несбалансированных транслокаций, а также относительно возможных клинических признаков "сбалансированных" транслокаций. Кроме теоретического интереса эти вопросы важны с точки зрения генетического консультирования - для оценки повторного риска.
2.2.2.2. Сегрегация и пренатальная селекция транслокаций: методологические аспекты
Проблема сегрегации и пренатальной селекции транслокаций служила предметом многих исследований, но результаты их оказались противоречивыми и в конечном счете не позволили дать общую картину. Совсем недавно многие из этих проблем были проанализированы и решены Шефером [501а]. Опишем вкратце ход его рассуждений и результаты анализа.
Транслокации относительно редки. Поэтому ни одна исследовательская группа не может накопить достаточно большой материал для окончательного вывода. Следовательно, необходимо суммировать данные разных авторов, опубликованные в литературе. Однако такие данные содержат много ошибок. Некоторые из них будут обсуждаться позже (разд. 3.3.4 и приложение 3). Например, все сибсы с аномальной хромосомой, как правило, учитываются только в том случае, если по крайней мере один из них поражен. Важно также и то, откуда отобраны сибства с одним пораженным: из общей популяции или только из небольшой ее части? Эти проблемы могут быть решены довольно просто, если мы имеем дело с наследственной болезнью, поскольку в этом случае семьи учитываются по одному пробанду, пораженному данной болезнью. С транслокациями дело обстоит сложнее, так как семьи могут быть учтены, например, по поводу повторных выкидышей или по пробанду, у которого обнаружена несбалансированная транслокация при рождении, или в результате пренатальной диагностики. Иногда семья оказывается в поле зрения исследователя по причине того, что в ходе популяционного исследования в ней обнаруживают носителя сбалансированной транслокации. Учитывая, что во многих публикациях отсутствует необходимая информация, полностью скорректировать все ошибки, конечно, невозможно, однако корректирующие процедуры Шефера являются на сегодняшний день оптимальными (см. приложение 3).
Данные, использованные для этого анализа. Исследование основано на результатах изучения 1050 семей с сегрегирующими транслокациями. Охвачено 2109 пар родителей и 4745 потомков. Кроме того, были суммированы данные о 556 случаях клинических проявлений у носителей сбалансированных транслокаций, о 814 пренатальных диагностических исследованиях и о 130000 индивидов, обследованных в ходе выполнения различных скринирующих программ. Это статистическое исследование позволило детализировать оценки повторного риска и фенотипические проявления у носителей сбалансированных и несбалансированных транслокаций. Для правильной интерпретации этих результатов необходимо наглядно представить себе последствия транслокаций во время мейоза.
Сегрегация транслокаций в первом мейотическом делении. В первом мейотическом делении хромосомы конъюгируют. Это относится и к транслоцированным хромосомным сегментам: они конъюгируют с гомологичными участками своих "партнеров", что приводит к образованию комплекса из четырех хромосом при реципрокной транслокации и комплекса из трех хромосом при робертсоновской перестройке. Как и в случае нормального мейоза, нити веретена фиксированы в центромерах и гомологичные хромосомы движутся к противоположным полюсам. В результате анафазного расхождения с равными вероятностями формируются четыре различных продукта деления (рис. 2.64):
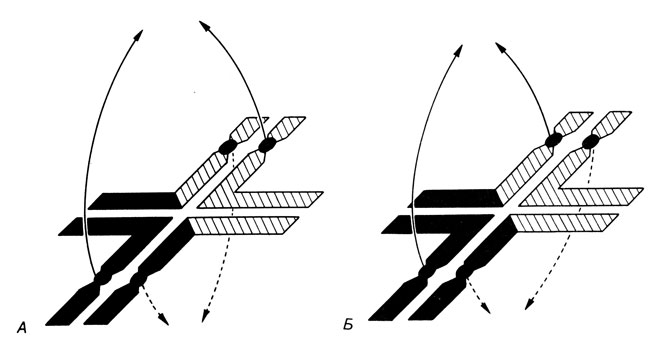
Рис. 2.64. Схематическое изображение транслокационного квадривалента, предполагающего альтернативное (А), или совместное-1 (Б) расхождение. Центрические сегменты длиннее, чем транслоцированные. Альтернативное расхождение дает нормальные и сбалансированные зиготы, при совместном расхождении образуются два типа несбалансированных зигот
1 и 2. Две нормальные хромосомы A1, B1 попадают в одну гаплоидную клетку (гамету), а две транслоцированные хромосомы A2, B2 - в другую гамету (альтернативное расхождение).
3 и 4. Одна нормальная и одна транспонированная хромосомы попадают в одну гамету (совместное 1-расхождение). В данном случае имеются две возможности: A1B2 или A2B1. Каждая из этих двух комбинаций имеет вероятность 0,25.
Вариант A1B1 кариотипически нормален, A2B2 - сбалансирован по транслокации, поскольку две хромосомы A2 и B2 только обменялись сегментами. Варианты A1B2 и A2B1 не сбалансированы по транслокации вследствие частичной трисомии. Кроме того, в результате этой хромосомной аберрации могут наблюдаться и другие, аномальные типы сегрегации. Например, гомологичные центромеры могут попадать иногда в одну гамету (совместное 2-расхождение; рис. 2.65), или в одну гамету попадают три центромеры, а в другую - только одна центромера (расхождение 3:1; рис. 2.66). Пренебрегая этими аномальными ситуациями, мы приходим к выводу, что среди потомков индивида со сбалансированной реципрокной транслокацией мы должны ожидать 50% носителей несбалансированной транслокации и, следовательно, с фенотипическими нарушениями; 25% будут иметь сбалансированную транслокацию и фенотипически нормальны; и 25% окажутся генотипически и фенотипически нормальными. Экспериментальные данные, однако, эти теоретически ожидаемые пропорции не подтверждают, причем количество детей с несбалансированными кариотипами оказывается намного меньше. Это может быть следствием многих причин: дополнительных аномалий в первом мейотическом делении, селекции в гаметогенезе против половых клеток с аномальным кариотипом, предпочтительного участия в оплодотворении нормальных и/или сбалансированных по транслокации половых клеток, а также селекции против анеуплоидных гамет на различных стадиях эмбрионального развития. Результаты статистического анализа могут дать ряд аргументов в пользу реального значения некоторых из этих механизмов.
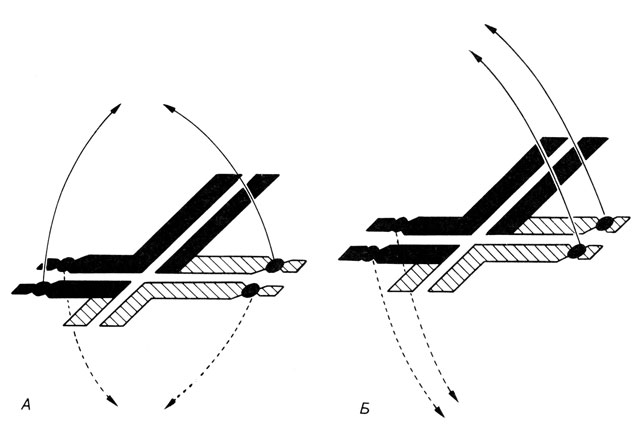
Рис. 2.65. Обе хромосомы, участвующие в транслокации - акроцентрические. Одно из спаривающихся плеч, которое не имеет центромеры, намного длиннее плеча с центромерой. В данном случае гомологичные центромеры могут (редко) попасть в одну и ту же дочернюю клетку (совместное 2-расхождение). Этот тип расхождения возможен также, если одна из двух хромосом является хромосомой 9
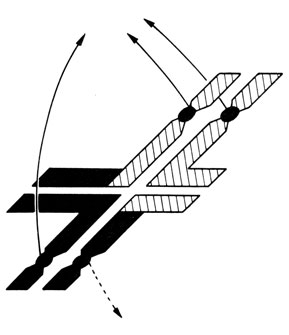
Рис. 2.66. Сегрегация 3:1 с образованием трисомии (или моносомии). Длина спаренных сегментов между центромерами достаточна для образования хиазм; хиазмы на фигуре справа не могут терминализоваться должным образом
Ожидаемые пропорции несбалансированных зигот. Если использовать указанные в приложении 3 поправки на ошибки выборки, то средний риск рождения потомков с несбалансированной транслокацией можно оценить в 7% для матерей, носителей сбалансированной транслокации, и в 3% для носителей-отцов. Эти значения риска были получены для всех носителей сбалансированной транслокации. Для носителей из семей, в которых несбалансированные по транслокации потомки были зарегистрированы уже ранее, риск для сибсов суммарно оказывается выше (14% от всех новорожденных для матерей и 8% для отцов); риск рождения несбалансированных по транслокации сыновей у отцов, носителей сбалансированной транслокации, особенно низок (5%). Если носителями являются матери, то среди всех несбалансированных по транслокации потомков 66% относится к типу "совместное-1", 3% - к типу "совместное-2" и 31% - к типу "3:1". Если носителями являются отцы, то 90% "несбалансированных" потомков будет типа "совместное-1", 3%-типа "совместное-2" и 8%-типа "3:1".
Для носителей транслокаций, выявленных по данным пренатальной диагностики, оценки риска таковы: 11,7% несбалансированных потомков в случае носителей-матерей и 12,1% для носителей-отцов. Низкая общая оценка (7% для носителей-матерей; 3% для отцов) связана с тем, что только около половины всех транслокаций могут проявиться в виде пороков развития при рождении, остальные приводят к внутриутробной гибели.
Для робертсоновских транслокаций с участием хромосомы 21 риск появления "несбалансированного" потомства составляет 13%, если носителем является мать, и 3%, если носитель - отец. С другой стороны у носителей транслокации DqDq риск рождения "несбалансированного" потомства практически нулевой. То же, по-видимому, справедливо и для транслокации Dq22q. Для генетического консультирования имеющиеся оценки риска в случае робертсоновских транслокаций достаточно надежны, но для реципрокных транслокаций они довольно грубые, поскольку получены при обобщенном подсчете многих типов транслокаций, в которых участвует много разных хромосом; можно ввести дополнительные поправки. Например, для носителя сбалансированной транслокации, который обследуется в связи с наличием у пробанда несбалансированной транслокации, риск выше, чем в случае, когда пробанд сам является носителем сбалансированной перестройки. Кроме того, должны приниматься во внимание специальные параметры, например такие, как длина хромосомы, участвующей в транслокации, и особенно размер трисомного (или моносомного) сегмента. В первом приближении можно считать, что чем больше этот сегмент, тем сильнее действует пренатальный отбор против анеуплоидной половой клетки или зиготы. Накапливается все больше данных в пользу предположения о том, что риск выше, если общая длина двух хромосомных сегментов, участвующих в дисбалансе, оказывается меньше 2% общей длины генома. Когда общая длина вовлеченных в транслокацию участков больше, вероятность ранней гибели "несбалансированных" зигот также увеличивается. Частичные моносомии имеют клинически более тяжелые последствия, чем частичные трисомии. В идеальном случае оценка риска должна быть основана на эмпирических данных об идентичной транслокации в той же самой семье [355]. Однако в большинстве случаев такие данные невозможно получить, так как существует слишком много различных транслокаций. Буэ [310], располагающий самым большим материалом по данной проблеме, указывает на то, что для любой робертсоновской или реципрокной транслокации характер выборки определяет величину повторного риска. Так, если сбалансированная транслокация обнаружена в ходе цитогенетического обследования по поводу повторных выкидышей, риск обнаружения плодов с несбалансированными транслокациями невелик (2%) (табл. 2.8). Наоборот, если выборка формировалась по аномальным детям, то риск рождения детей с несбалансированными перестройками составляет почти 20%. Такой анализ способствует пониманию особенностей сегрегации транслокаций в первом мейотическом делении у человека. В общем, сегрегация, по-видимому, происходит вполне правильно: гомологичные центромеры действительно мигрируют к противоположным полюсам, что ведет либо к "альтернативной", либо к "совместной-1" сегрегации. В виде исключения иногда может происходить сегрегация по типу "совместная-2" или "3:1"; последняя происходит в основном в тех случаях, когда маленькая хромосома остается неспаренной. У мужчин это часто ведет к нарушениям мейоза (рис. 2.66). Отбор против несбалансированных гамет происходит у мужчин, но не у женщин. Отбор против несбалансированных зигот зависит от типа и масштабов хромосомного дисбаланса; очень крупный дисбаланс будет приводить к ранней и нераспознаваемой гибели эмбрионов. Умеренный дисбаланс приведет к диагностируемому спонтанному аборту. Частота выкидышей увеличивается особенно в том случае, если носителем является мать. Это может быть связано с отсутствием у женщин гаметического отбора.
![Таблица 2.8. Выявление транслокаций [310]](pic/000078.jpg)
Таблица 2.8. Выявление транслокаций [310]
В прошлом предполагалось, что наличие в кариотипе транслокации может повышать вероятность нарушения сегрегации других хромосом. Например, регулярная трисомия 21, согласно данным некоторых авторов, чаще наблюдалась в потомстве матерей, которые были носителями сбалансированной транслокации. Однако тщательный анализ конкретных материалов не подтвердил это предположение.
Сегрегация кариотипически нормальных и сбалансированных зигот. Как уже указывалось, альтернативная сегрегация теоретически должна вести к появлению кариотипически нормальных и "сбалансированных" зигот в равных отношениях (50:50). На практике в большинстве сообщений приводятся данные о небольшом избытке "сбалансированных" зигот. Анализ Шефера [501а] показал, что этот факт отражает биологическую реальность. Подобное явление наблюдается у сыновей, отцы которых - носители сбалансированной транслокации, и особенно явно у сыновей, отцы которых несут транслокацию 13ql4.
Фенотипические отклонения у носителей сбалансированных транслокаций. Носители сбалансированных транслокаций имеют полный набор генетического материала и, следовательно, должны быть фенотипически нормальными. Как правило, это подтверждается на практике. Однако во многих сообщениях приводятся данные о небольшом повышении частоты пороков развития, умственной отсталости и малых врожденных дефектов. Анализ этих данных показал, что множественные пороки развития и умственная отсталость редко встречаются среди носителей сбалансированных транслокаций, но чаще, чем среди кариотипически нормальных людей (табл. 2.9). Как правило, такие клинические находки встречаются при спорадических или семейных транслокациях Dq21q: в этих семьях можно предполагать наличие необнаруженных мозаиков. При спорадических транслокациях появление аномального фенотипа можно объяснить разрывом в гене или его изъятием из функционально значимого окружения (эффект положения). Большинство носителей семейных транслокаций клинически здоровы. Отбор против несбалансированных зигот ведет к увеличению частоты выкидышей; среди супружеских пар с привычным невынашиванием частота носителей транслокаций выше. Бесплодие обычно наблюдается у носителей-мужчин; а у женщин - при X-аутосомных транслокациях. Однако эти данные не должны затмевать тот факт, что подавляющее большинство носителей семейных и спорадических транслокаций имеют нормальный фенотип.
![Таблица 2.9. Частота (на 1000 индивидов) сбалансированных транслокаций в различных группах [501а]](pic/000079.jpg)
Таблица 2.9. Частота (на 1000 индивидов) сбалансированных транслокаций в различных группах [501а]
Приведенные данные о транслокациях у человека имеют значение для оценки частоты мутаций, а также для оценки приспособленности, силы отбора и для теории эволюции.
|
ПОИСК:
|
При использовании материалов активная ссылка обязательна:
http://genetiku.ru/ 'Генетика'